 2015, Vol. 34
2015, Vol. 34文章信息
- 杨冰, Pignatello Joseph J, 曲东
- YANG Bing, Pignatello Joseph J, QU Dong
- 利用固体类Fenton试剂降解五氯酚的探讨
- Decontamination of Pentachlorophenol Using Solid Fenton-like Reagents
- 农业环境科学学报, 2015, 34(10): 1914-1920
- Journal of Agro-Environment Science, 2015, 34(10): 1914-1920
- http://dx.doi.org/10.11654/jaes.2015.10.011
-
文章历史
- 收稿日期: 2015-07-10





2. Department of Environmental Sciences, The Connecticut Agricultural Experiment Station, 123 Huntington Street, P.O. Box 1106, New Haven, Connecticut 06504-1106, United States
2. Department of Environmental Sciences, The Connecticut Agricultural Experiment Station, 123 Huntington Street, New Haven, Connecticut 06504-1106, United States
Fenton反应通过Fe2+和H2O2反应生成强氧化剂羟基自由基(·OH),能够快速化分解大多数有机物[1],广泛用于去除水体有机污染物[2, 3, 4]。但在污染土修复中,由于土壤组成的复杂性和低传质性,要求Fenton试剂有足够的持续效性来增加扩散半径[5]。在微生物作用、土壤颗粒表面催化、土壤中过渡金离子催化下,H2O2极易发生无效分解,生成O2和H2O[6, 7, 8]。同时,传统enton反应要求最佳pH在3~4[2, 5],而实际土壤由于具有较强的pH缓冲能力难满足这一要求。因此,传统Fenton试剂(FeSO4和H2O2)在土壤有机污染修中受到很大限制。
已有研究在均相体系中使用固体过氧化物代替H2O2用于(类)Fenton反应,并有效降解有机污染物。如在含Fe3+-EDTA的水溶液(pH 6~9)中,使用过氧钙(CP)比H2O2对四氯乙烯(TCE)的矿化率更高,原因是CP缓慢水解能可控释放出H2O2,减少了H2O2的无效分解[9]。在过碳酸钠(SPC)与FeSO4或添加螯合剂的Fe2+体系中,TCE[10, 11]和苯[12]能得到有效地降解。但在非均相系,如污染土壤中,使用固体过氧化物基于Fenton化学降解有机物的研究还少。同时,稳定的Fe(Ⅱ)来源是保证Fenton反应可持续性的另一重要因素可通过循环使用Fe(Ⅲ)产物或者额外提供Fe(Ⅱ)源(如铁矿物)来实现这种方法通常要求反应体系有足够的酸度[2, 7],或者需要添加螯合剂使FeⅢ)处于溶解状态[13, 14]。已报道用来催化Fenton反应的天然铁矿物主要有铁矿、 水铁矿、针铁矿、纤铁矿、黄铁矿及磁铁矿[15, 16, 17, 18]。而人工合成的米级铁氧化物,由于其易分散、稳定性好、适应的pH范围宽,具有比天然矿更好的催化性能,已有诸多研究将其用于类Fenton反应[19, 20, 21]。
本研究以五氯酚(PCP)污染的石英砂模拟污染土壤,选用目前研究报道较多并业化大量生产的四种固体过氧化物——CP、SPB(过硼酸钠)、SPC、UHP(过化尿素),以及人工合成的纳米级Fe3(PO4)2作为固体类Fenton试剂,与传的H2O2和FeSO4组合,探究不同试剂用量、H2O2和不同固体过氧化物与FeSO4Fe3(PO4)2组合对PCP的降解能力差异,阐明影响固体类Fenton试剂活性成有效性和持续性的主要因素,为进一步提高固体类Fenton试剂在非均相体系降解有机污染物的效率提供必要的依据。
1 材料与方法 1.1 实验试剂与仪器 1.1.1 主要试剂SPC、SPB、UHP、(NH4)2Fe(SO4)2·6H2O、PCP、FeSO4、K2HPO4·3H2O均为析纯,H2O2(W/W=30%)、CP[75% CaO2/25% Ca(OH)2]均购自美国Sigma-ldrich公司。NH4VO3(钒酸铵,分析纯)购自美国Matheson Coleman & Bell司,KI(分析纯)和石英砂(40~100目)购自美国Acros Organics公司,甲(色谱纯)购自美国Fisher Chemical公司。实验用水均为超纯水。
1.1.2 仪器高效液相色谱仪(安捷伦R1100),色谱柱4.6×150 mm XDB-C18柱,紫外检测Diode-array。 紫外可见分光光度计Hewlett-Packard 8452A。pH计Orion 20A,玻璃电极。
1.2 制备五氯酚污染的石英砂于通风厨内,将500 g石英砂置于2 L烧杯中,添加5 g·L-1 PCP的丙酮储备液5 mL,并额外分次添加共100 mL丙酮,添加过程中保持搅拌,直到丙酮挥发完。此方法得到的石英砂中PCP含量约为250 mg·kg-1,折合0.94 mmol·kg-1。
1.3 制备纳米级Fe3(PO4)2采用Scaccia等[22] 报道的方法合成纳米级Fe3(PO4)2,颗粒物粒径为80~200 m。室温下,将250 mL 0.1 mol·L-1(NH4)2Fe(SO4)2溶液(N2除氧)缓慢入含有250 mL 0.067 mol·L-1 K2HPO4溶液的1 L三角瓶中,磁力搅拌器保持拌,pH计连续监测,瓶内上方持续通入N2减少空气影响。使用1∶1稀释后的氨水调节pH至6.5,白蓝色沉淀即出现,保持pH 6.5约10 min,撤去pH计,停搅拌,Parafilm膜封口,静置7 d,瓶内溶液和沉淀使用0.45 μm膜滤器过滤高纯水多次冲洗,所得沉淀60 ℃下真空干燥24 h,呈均匀粉末状,于干燥试保存待用。
1.4 实验设置实验采用10 mL带盖玻璃试管为反应容器,以聚四氟乙烯内衬硅胶垫密封。依次入1.0 g PCP污染的石英砂,不同用量的FeSO4或合成的Fe3(PO4)2和高纯水轻轻晃动摇匀后,加入一定量的固体过氧化物或H2O2,快速旋紧盖子,晃动反应开始计时。反应试管固定于垂直旋转转子上,转速60 r·min-1,置于(0±0.2)℃黑暗恒温培养箱中。按设定的时间随机取样测定PCP和H2O2含量,个重复。不同处理及试剂添加量如下,控制反应体系液体总体积为1 mL,则CP浓度为0.94 mmol·L-1。
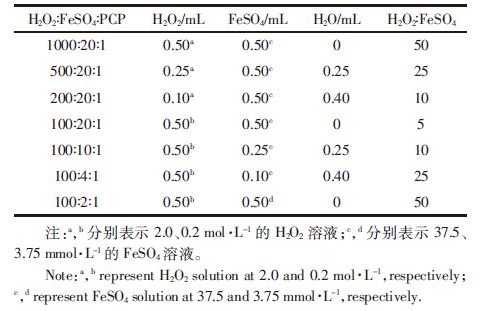
1.4.1 H2O2和FeSO4比例H2O2∶FeSO4∶PCP的摩尔比和试剂添加量如表 1。
 |
以铁源可给出的最大量Fe2+计算,使用H2O2∶Fe2+∶PCP摩尔比为100∶10∶1。剂添加量为1.0 mL H2O2(0.1 mol·L-1)+ 5.0 mg Fe3(PO4)2。
1.4.3 固体过氧化物和FeSO4或Fe3(PO4)2以过氧化物和铁源可给出的最大量H2O2和Fe2+计算,使用H2O2∶Fe2+∶PCP摩尔为100∶10∶1。固体过氧化物的添加量分别为9.0 mg CP、20.0 mg UHP、9.4 g SPB,12.8 mg SPC。铁源添加为1 mL FeSO4溶液(9.4 mmol·L-1),或5.0 g Fe3(PO4)2+1.0 mL高纯水。
1.5 测定项目及方法 1.5.1 PCP测定添加4 mL 68%甲醇水溶液于反应试管,避光振荡浸提24 h,1800 r·min-1离心5 min,上层液过滤(0.2 μm),滤液待测。PCP采用高效液相色谱(安捷伦1100)测定,流动相为甲醇∶乙酸溶液(1%)=85∶15,流速1 mL·min-1,柱25 ℃,检测波长304 nm,PCP保留时间约6 min。本实验石英砂中PCP回收率94.96%±0.54%(n=6),PCP在石英砂中最低检测限(S/N=10)为0.4 mg·kg-1。
1.5.2 H2O2含量测定添加4 mL高纯水于反应试管,旋紧盖快速摇动,1800 r·min-1离心10 min,上液过滤(0.2 μm),滤液待测。H2O2的测定采用Nogueira等[23]报道的方法基本原理为H2O2和NH4VO3在酸性条件下反应生成橙红色VO23+阳离子,测定其450 nm的吸光值来计算H2O2含量。标准工作曲线由H2O2溶液与NH4VO3反应得,标准溶液H2O2含量由碘量法测定。
1.5.3 活性过氧化物的含量测定固体过氧化物中活性过氧化物的含量采用Goi等[24]报道的方法测定。取一定量固体过氧化物溶解于含6%盐酸和14%磷酸的混合酸溶液,以0.5 mol·L-1的高酸钾滴定,测得的活性过氧化物含量以“mmol H2O2·g-1固体”计,在CP、PB、SPC和UHP 中分别为(10.79±0.08)、(9.53±0.06)、(8.41±0.09)(10.39±0.02)mmol·g-1,与试剂瓶上标注的活性氧或H2O2含量范围相符.
1.5.4 pH测定添加4 mL高纯水于反应试管,快速摇匀,使用Orion 720A pH计测定,所得pH经正体积变化导致的差异后为反应体系pH。
2 结果与分析 2.1 H2O2和FeSO4比例对PCP降解的影响不同摩尔浓度比的H2O2和FeSO4对石英砂中PCP的降解如图 1所示。在FeSO4∶PCP 20∶1的处理中(图 1a),H2O2用量在50倍到5倍于FeSO4时,PCP降解速率在 h内随H2O2用量减少略有降低;但降解率差异不大,6 h后为74.6%~79.9%,24 后为83.3%~84.0%。保持H2O2∶PCP为100∶1(图 1b),FeSO4用量为H2O2的/5~1/50时,PCP降解速率在6 h内随FeSO4用量减少有明显降低;在6 h降解率74.6%~28.0%,但在24 h后降解率较为接近,为83.5%~78.0%。在只添加FeSO4H2O2的对照中,24 h内PCP无明显降解。在H2O2∶FeSO4∶PCP =100∶20∶1和00∶10∶1的处理中,6 h内PCP降解率相当(约75%)。

|
| 图 1 不同添加比例的H2O2∶FeSO4∶PCP对PCP的降解 Figure 1 PCP degradation in treatments with H2O2∶FeSO4∶PCP at different molar ratio |
上结果说明在FeSO4∶PCP=20∶1时,H2O2使用量(5~50倍于Fe2+)不是限制子;而在固定H2O2∶PCP=100∶1时,Fe2+添加量(为H2O2的1/5~1/50)为影PCP降解速率的限制因子,但对最终的降解率贡献不大。综合考虑试剂用量和CP降解率,以下实验采用H2O2∶FeSO4∶PCP为100∶10∶1。
图 1所有处理中,PCP降解率(24 h)最高值在84%左右。在H2O2∶FeSO4∶CP=100∶10∶1体系中,监测PCP和H2O2浓度变化,24 h后加入新鲜H2O2和eSO4,以期降解剩余PCP,结果如图 2所示。反应初期,H2O2和PCP快速减少,6 时剩余H2O2仅为5%,PCP降解率达到约80%。24 h时已检测不到H2O2,此时PCP解率比6 h略微增加(81.5%),24~29 h PCP几乎无减少,与H2O2耗竭有关。在24 h时添加与初始添加量相当的H2O2和FeSO4后,H2O2在5 h内(24~29 h)次耗尽,PCP降解仅增加2%,仍有16%左右的PCP不能被降解,与图 1中情形一。结果表明,PCP的降解与H2O2密切相关,在石英砂中有约16%的PCP不能被本究的方法降解。

|
|
PCP*代表在24 h添加新鲜H2O2和FeSO4后PCP的变化 PCP* represents PCP change after adding fresh H2O2 and FeSO4 at 24 h 图 2 H2O2∶FeSO4∶PCP =100∶10∶1时体系PCP和H2O2的变化 Figure 2 Changes of PCP and H2O2 content at H2O2∶FeSO4∶PCP of 100∶10∶1 |
以固体过氧化物为H2O2来源和FeSO4为铁源时对PCP的降解如图 3所示。无FeSO4加时,SPB、SPC、UHP、H2O2在48 h内对PCP无明显降解,而CP对PCP在24 h和8 h内的降解率分别为10.4%和13.4%。同时添加过氧化物和FeSO4时,SPB、SPCUHP、H2O2在24 h内对PCP的降解分别为14.6%、17.3%、 86.5%、83.8%,但在4~48 h基本无增加。同时添加CP和FeSO4时,PCP在24 h和48 h内持续降解,分为22.0%和25.9%。使用CP时,添加与不添加FeSO4的处理中,PCP降解率在4~48 h分别增加3.9%和3.0%,二者较为接近,PCP持续降解可能主要由CP引起而添加FeSO4引起的Fenton反应贡献较小。

|
| 图 3 不同过氧化物与FeSO4组合对PCP的降解(P<0.05) Figure 3 PCP degradation in treatments with different peroxides in combination with FeSO4(P<0.05) |
在本研究体系中,SPB、SPC、UHP均能较快地溶解,而CP有大部分无法解,仍以固体粉末形态分布在溶液中或是与石英砂相混。在CP、SPB、SPC处中,溶液pH快速升至12.2、10.4、11.7,有FeSO4存在时,红褐色沉淀快速出,表明有Fe(OH)3或Fe2O3生成,并附着在试管壁和石英砂中。在HP、UHP处中,有FeSO4存在时,pH在2~3之间,溶液呈浅黄绿色,表明Fe2+和FFe3+共同在。
2.3 CP对PCP降解持续性的影响为进一步探明CP对PCP降解的持续性,在添加或不添加FeSO4的处理中对PCP在30 内的降解进行监测(图 4)。PCP快速降解主要在前4 d,降解率分别为18.7%(P)和26.5%(CP+FeSO4)。在4~29 d,CP+FeSO4处理中PCP降解率增幅小于3%表明体系能降解PCP的活性成分在第4 d后基本耗尽。而在CP处理中无FeSO4添时,PCP在4~29 d仍缓慢持续减少,降解率增幅为8.6%,表明体系仍有部分活氧化物持续作用。

|
| 图 4 CP和CP+FeSO4对PCP的降解 Figure 4 PCP degradation in treatment with CP or CP+FeSO4 |
H2O2和纳米Fe3(PO4)2对PCP在16 d内的降解如图 5所示。在反应开始1 h内,CP降解率为5.6%,而后降解明显变慢,1~24 h降解率增加为5.4%。PCP在实验间持续减少,表明活性成分一直存在。第16 d时,PCP降解率为55.6%,同时得剩余H2O2为54.2 mmol·L-1,约为初始添加量一半,表明体系H2O2充足,致PCP降解缓慢的原因可能是活性Fe2+不足。在不同反应时间段0~1 h、1~2 h2~6 h、6~48 h、48 h~9 d、9~16 d,PCP平均降解速率分别为13.6、4.5、.2、0.16、0.14、0.46 mg·kg-1·h-1。可见,在反应的前6 h内PCP的降解速急剧变慢,而在6 h~9 d较为接近,但9~16 d的平均速率有所增加,为2~9 d3.3倍。实验期间体系的pH保持在4~5之间,溶液颜色无明显变化,无其他可沉淀生成,Fe3(PO4)2仍以固体形式存在于溶液中或是与石英砂相混,表明e3(PO4)2在整个反应过程中较为稳定。

|
| 图 5 H2O2+Fe3(PO4)2对PCP的降解 Figure 5 PCP degradation in treatment with H2O2 plus Fe3(PO4)2 |
以固体过氧化物作为H2O2来源与以Fe3(PO4)2为Fe(Ⅱ)源对PCP在20 d内的解如图 6所示。PCP在反应初期5 d内快速降解,而后在5~20 d降解变缓,与.4.1中H2O2和Fe3(PO4)2对PCP的降解相似。在UHP处理中,5 d内PCP降解率0.7%,而后缓慢增加,在7、14、20 d时分别为23.9%、30.4%、37.0%。在CP、PB、SPC处理中,PCP降解率在前5 d分别为20.7%、8.9%、4.3%,5~20 d内分别加9.4%、4.8%、4.2%。无过氧化物添加时,Fe3(PO4)2对PCP无降解作用,英砂中PCP本身在20 d内也无降解。

|
| 图 6 固体过氧化物+Fe3(PO4)2对PCP的降解 Figure 6 PCP degradation in treatments with solid peroxides and Fe3(PO4)2 |
根据Fenton反应中导致H2O2分解为O2的自由基机理[2],本实验体系中可能发生主要反应如方程(1)~(6)。可见,在Fenton反应中H2O2和Fe(Ⅱ)均可能有机物竞争消耗·OH,反应(3)、(4)、(5)。过量的H2O2与·OH反应生氧化活性较低的HOO·,同时HOO·与HOO·反应生成H2O2和O2,导致增加H2O2量却不能提高甚至降低有机物降解效率。这与我们在2.1中观察到的结果相符当固定Fe2+使用量,H2O2∶PCP在100~1000之间时,PCP降解差异并不明显。固定H2O2使用量时,Fe2+∶H2O2在1/5~1/50之间,PCP的降解速率随初始Fe2+量的增加而显著增加。这是因为当H2O2相对于Fe2+过量时,初始产生的·OH与Fe2+为等摩尔量[反应(1)]。
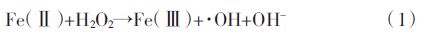
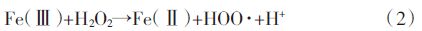

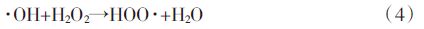
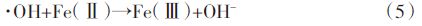
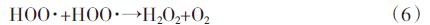
燕启社等[25]在研究Fenton反应去除土壤中有机污染物时采用H2O2∶FeSO4为101,污染物去除率在120 min内能达到65%~89%,与本实验体系中采用H2O2∶eSO4∶PCP最佳比100∶10∶1一致,但均比水处理中常使用的H2O2∶Fe(Ⅱ)围100~1000[2]更低。这是由于PCP负载在石英砂上,处于非均相体系,其中量的H2O2对·OH的竞争消耗比在均相体系更严重。
3.2 过氧化物与FeSO4组合添加SPB、SPC于石英砂和水1∶1的体系中,SPB、SPC均能快速溶解并释放出2O2[26],且溶液pH快速升高至碱性(pH 10~12),导致FeSO4以及H2O2和Fe2+应生成的FFe3+形成Fe(OH)2和Fe(OH)3沉淀,二者在Fenton反应中都是活很低的铁源[2],无法为反应持续提供Fe2+。因此在添加FeSO4和SPB或SPC的理中,仅观察到PCP在24 h内的一次性明显降解,而后在24~48 h降解无明显加(图 3)。Miao等[11] 使用低浓度等摩尔比的SPC和FeSO4,在水溶液中初pH约6.5,能高效降解TCE。而在添加H2O2、UHP的处理中,pH保持在2~3之间Fe2+和FFe3+能够共同存在,Fenton反应能较好地持续进行,直到过氧化物在4 h内消耗完(图 2)。添加CP的处理中,CP仅有小部分溶解,但溶液pH快速升12以上,使Fe(Ⅱ)源失活,同时CP缓慢溶解持续释放出H2O2,为碱催化H2O2降解PCP提供了可能[27, 28]。因此在CP以及CP+FeSO4的处理中观察到PCP有续的降解(图 3和图 4)。可见,以FeSO4为铁源时,添加固体过氧化物对体系H的改变,极大程度上影响到Fe2+的有效性,进而影响Fenton反应活性和持续。
3.3 过氧化物与Fe3(PO4)2组合使用固体铁源时,颗粒物表面催化作用以及溶液中可用Fe(Ⅱ)的含量均是影Fenton反应效率的重要因素[16]。H2O2与Fe3(PO4)2组合中,Fe3(PO4)2粒表面Fe(Ⅱ)活性催化位点在反应初期快速催化H2O2产生·OH氧化降解PCP图 5),同时Fe(Ⅱ)变成Fe(Ⅲ)。但Fe(Ⅲ)矿物是低效的Fenton铁源 [14],且与H2O2反应再生成Fe(Ⅱ)较缓慢,致使颗粒物表面催化的Fenton反因Fe(Ⅲ)和Fe(Ⅱ)循环的断链而减缓或终止。Fe3(PO4)2在水溶液中(5 ℃)溶度积常数为1.71×10-36,溶解平衡时Fe2+浓度为8.3×10-8 mol·L-1 [29]。在本研究中尽管固体Fe3(PO4)2添加量为1.1×10-2 mol·L-1,但Fe3(PO4)2有限溶解使溶液中Fe2+保持低浓度(≤8.3×10-8 mol·L-1),能缓慢但持续催化H2O2氧化降解PCP。在第16 d测得溶液中H2O2仍有(54.2±.1)mmol·L-1,远高于(>6.5×105倍)Fe2+浓度,表明在实验期间H2O2处于量过量状态,对生成的·OH有较大消耗作用。随反应进行,体系H2O2含量减,对·OH的竞争消耗也随之减弱,同时部分PCP从石英砂中解吸使溶液中PCP量增加,因此在实验后期(9~16 d) PCP平均降解速率反而有所增加。这说在铁源和污染物均处于非均相体系时,H2O2使用量还应该再降低,可以通过量多次添加提高对有机物的去除效率[30]。
在固体过氧化物与Fe3(PO4)2的组合中(图 6),当致碱性过氧化物CP、SPB、PC添加到水溶液中,溶液中Fe2+和Fe3+与OH-结合生成Fe(OH)2(Ksp=8×10-16)和Fe(OH)3(Ksp=1×10-36)[31],二者在碱性溶液中的溶解度远低于Fe3(PO4)2。同时,生成的Fe(OH)2和Fe(OH)3覆盖在Fe3(PO4)2颗粒表可能阻碍Fe2+的溶解,覆盖在石英砂表面阻碍PCP溶解或阻碍PCP与生成的活氧化物接触。而在UHP或H2O2 + Fe3(PO4)2的处理中,pH稳定在4~5,溶液Fe2+含量比CP、SPB、SPC处理高出很多,因此在5~20 d内CP、SPB、SPC处理PCP降解率增幅显著低于UHP和H2O2处理。
4 结论(1)固体类Fenton试剂在20 d内对石英砂体系中PCP的去除率为8.6%~55.6%,传统Fenton试剂(24 h内达84%)相比其去除率较低,但试剂的持续有效时间到较大幅度延长,由小于24 h增加至20 d以上,故在土壤污染修复中具有更潜力。
(2)致碱性过氧化物(CP、SPB、SPC)作为类Fenton试剂时需要配合pH缓冲剂提高对有机物的降解率,而UHP在一定程度上可以替代H2O2。
(3)固体铁源Fe3(PO4)2溶解度过低,限制了其对PCP的降解速率,但其能够定提供Fe2+,可以持续降解PCP。使用溶解度比Fe3(PO4)2稍高且稳定的固铁源,有望提高有机物的降解速率和效率。
| [1] | Buxton G V, Greenstock C L, Helman W P, et al. Critical review of rate constants for reactions of hydrated electrons, hydrogen-atoms and hydroxyl radicals(·OH/·0-) in aqueous-solution[J]. Journal of Physical and Chemical Reference Data, 1988, 17(2):513-886. |
| [2] | Pignatello J J, Oliveros E, MacKay A. Advanced oxidation processes for organic contaminant destruction based on the Fenton reaction and related chemistry[J]. Critical Reviews in Environmental Science and Technology, 2006, 36(1):1-84. |
| [3] | Wang J L, Xu L J. Advanced oxidation processes for wastewater treatment:Formation of hydroxyl radical and application[J]. Critical Reviews in Environmental Science and Technology, 2012, 42(3):251-325. |
| [4] | Bautista P, Mohedano A F, Casas J A, et al. An overview of the application of Fenton oxidation to industrial wastewaters treatment[J]. Journal of Chemical Technology and Biotechnology, 2008, 83(10):1323-1338. |
| [5] | Watts R J, Teel A L. Chemistry of modified Fenton′s reagent(catalyzed H2O2 propagations-CHP) for in situ soil and groundwater remediation[J]. Journal of Environmental Engineering, 2005, 131(4):612-622. |
| [6] | Petigara B R, Blough N V, Mignerey A C. Mechanisms of hydrogen peroxide decomposition in soils[J]. Environmental Science & Technology, 2002, 36(4):639-645. |
| [7] | Baciocchi R, Boni M R, D′Aprile L. Hydrogen peroxide lifetime as an indicator of the efficiency of 3-chlorophenol Fenton′s and Fenton-like oxidation in soils[J]. Journal of Hazardous Materials, 2003, 96(2/3):305-329. |
| [8] | Watts R J, Foget M K, Kong S H, et al. Hydrogen peroxide decomposition in model subsurface systems[J]. Journal of Hazardous Materials, 1999, 69(2):229-243. |
| [9] | Northup A, Cassidy D. Calcium peroxide(CaO2) for use in modified Fenton chemistry[J]. Journal of Hazardous Materials, 2008, 152(3):1164-1170. |
| [10] | Miao Z W, Gu X G, Lu S G, et al. Mechanism of PCE oxidation by percarbonate in a chelated Fe(Ⅱ)-based catalyzed system[J]. Chemical Engineering Journal, 2015, 275:53-62. |
| [11] | Miao Z W, Gu X G, Lu S G, et al. Perchloroethylene(PCE) oxidation by percarbonate in Fe2+-catalyzed aqueous solution:PCE performance and its removal mechanism[J]. Chemosphere, 2015, 119:1120-1125. |
| [12] | Fu X R, Gu X G, Lu S G, et al. Benzene depletion by Fe2+-catalyzed sodium percarbonate in aqueous solution[J]. Chemical Engineering Journal, 2015, 267:25-33. |
| [13] | Pignatello J J, Day M. Mineralization of methyl parathion insecticide in soil by hydrogen peroxide activated with iron(Ⅲ)-NTA or -HEIDA complexes[J]. Hazardous Waste & Hazardous Materials, 1996, 13(2):237-244. |
| [14] | Sun Y F, Pignatello J J. Chemical treatment of pesticide wastes-evaluation of Fe(Ⅲ) chelates for catalytic hydrogen-peroxide oxidation of 2, 4-D at circumneutral pH[J]. Journal of Agricultural and Food Chemistry, 1992, 40(2):322-327. |
| [15] | Watts R J, Udell M D, Kong S H, et al. Fenton-like soil remediation catalyzed by naturally occurring iron minerals[J]. Environmental Engineering Science, 1999, 16(1):93-103. |
| [16] | Matta R, Hanna K, Chiron S. Fenton-like oxidation of 2, 4, 6-trinitrotoluene using different iron minerals[J]. Science of the Total Environment, 2007, 385(1/3):242-251. |
| [17] | Huang H H, Lu M C, Chen J N. Catalytic decomposition of hydrogen peroxide and 2-chlorophenol with iron oxides[J]. Water Research, 2001, 35(9):2291-2299. |
| [18] | Kong S H, Watts R J, Choi J H. Treatment of petroleum-contaminated soils using iron mineral catalyzed hydrogen peroxide[J]. Chemosphere, 1998, 37(8):1473-1482. |
| [19] | Ardo S G, Nelieu S, Ona-Nguema G, et al. Oxidative degradation of nalidixic acid by nano-magnetite via Fe2+/O2-mediated reactions[J]. Environmental Science & Technology, 2015, 49(7):4506-4514. |
| [20] | 裴 欢, 毛 飞, 司友斌. 纳米铁氧化物催化类Fenton反应降解抗生素磺胺[J]. 农业环境科学学报, 2015, 34(7):1356-1362. PEI Huan, MAO Fei, SI You-bin. Degradation of antibiotic sulfanilamide in aqueous solution via a heterogeneous Fenton-like reaction catalyzed by nano-iron oxides[J]. Journal of Agro-Environment Science, 2015, 34(7):1356-1362. |
| [21] | Pereira M C, Oliveira L C A, Murad E. Iron oxide catalysts:Fenton and Fenton-like reactions: A review[J]. Clay Minerals, 2012, 47(3):285-302. |
| [22] | Scaccia S, Carewska M, Di Bartolomeo A, et al. Thermoanalytical investigation of nanocrystalline iron(Ⅱ) phosphate obtained by spontaneous precipitation from aqueous solutions[J]. Thermochimica Acta, 2003, 397(1/2):135-141. |
| [23] | Nogueira R F, Oliveira M C, Paterlini W C. Simple and fast spectrophotometric determination of H2O2 in photo-Fenton reactions using metavanadate[J]. Talanta, 2005, 66(1):86-91. |
| [24] | Goi A, Viisimaa M, Trapido M, et al. Polychlorinated biphenyls-containing electrical insulating oil contaminated soil treatment with calcium and magnesium peroxides[J]. Chemosphere, 2011, 82(8):1196-1201. |
| [25] | 燕启社, 孙红文, 周长波, 等. 类Fenton氧化在污染土壤修复中的应用[J]. 生态环境, 2008, 17(1):216-220. YAN Qi-she, SUN Hong-wen, ZHOU Chang-bo, et al. Application of Like-Fenton oxidation on remediation of contaminated soil[J]. Ecology and Environment, 2008, 17(1):216-220. |
| [26] | McKillop A, Sanderson W R. Sodium perborate and sodium percarbonate:Cheap, safe and versatile oxidising agents for organic synthesis[J]. Tetrahedron, 1995, 51(22):6145-6166. |
| [27] | Arienzo M. Degradation of 2, 4, 6-trinitrotoluene in water and soil slurry utilizing a calcium peroxide compound[J]. Chemosphere, 2000, 40(4):331-337. |
| [28] | Katafias A, Lipińska M, Strutyński K. Alkaline hydrogen peroxide as a degradation agent of methylene blue—kinetic and mechanistic studies[J]. Reaction Kinetics, Mechanisms and Catalysis, 2010, 101(2):251-266. |
| [29] | Al-Borno A, Tomson M B. The temperature dependence of the solubility product constant of vivianite[J]. Geochimica et Cosmochimica Acta, 1994, 58(24):5373-5378. |
| [30] | Chu W, Chan K H, Kwan C Y, et al. Degradation of atrazine by modified stepwise-Fenton′s processes[J]. Chemosphere, 2007, 67(4):755-761. |
| [31] | Stumm W, Lee G F. Oxygenation of ferrous iron[J]. Industrial & Engineering Chemistry, 1961, 53(2):143-146. |