 2016, Vol. 35
2016, Vol. 35文章信息
- 李龙山, 倪细炉, 李昌晓, 李健
- LI Long-shan, NI Xi-lu, LI Chang-xiao, LI Jian
- 生活污水对土壤及湿地植物根际细菌群落的影响
- Impacts of domestic sewage on community structure and diversity of bacteria in the soil and rhizospheres of five wetland plants
- 农业环境科学学报, 2016, 35(11): 2163-2170
- Journal of Agro-Environment Science, 2016, 35(11): 2163-2170
- http://dx.doi.org/10.11654/jaes.2016-0506
文章历史
- 收稿日期: 2016-04-13






2. 宁夏鼎实生物鉴定中心, 银川 750002 ;
3. 宁夏银川城市森林生态系统国家定位观测研究站, 宁夏 银川 750004
2. Ningxia Ding-Shi Bioassay Center, Yinchuan 750002, China ;
3. Yinchuan Urban Forest Ecosystem Research Station of State Forestry Administration, Yinchuan 750004, China
湿地处于陆地和水域的交汇处,是陆地和水生生态系统的中间地带,因其具有强大的净化能力受到科学家和工程师青睐,通过构建人工湿地用于各类污水的净化[1-3]。人工湿地净化污水的机制比较复杂,包括物理沉降、化学吸附、离子转换、植物吸收和微生物降解等过程[4]。探究污水净化过程中湿地微生物群落结构变化,了解土壤—水—植物湿地系统中所发生的生化反应过程,对人工湿地的设计、维护和运行具有重要参考价值。目前,对人工湿地微生物的研究主要集中在以下几个方面:一是人工湿地中植物对微生物的影响。如赵庆节[5]利用DGGE技术研究显示湿地植物对土壤中微生物的纵向分布上有较大影响,但不起决定作用。李鑫等[6]研究发现不同植物对土壤中微生物的影响作用不同。二是湿地微生物与污染物去除之间的关系研究[7]。杜刚等[4]研究发现微生物数量与氨氮、总磷和CODMn的去除率不相关,但与总氮去除率显著正相关。三是人工湿地处理污水后污水中微生物变化的研究。郭建国等[8]研究发现人工湿地处理造纸厂废水后,废水中的病原微生物数量明显降低。
大部分微生物在实验室条件下是不能培养的,传统的微生物培养方法只能培养1%~10%的微生物类群[9],所以传统的培养方法明显低估了环境微生物的多样性。PCR-DGGE技术不需要进行微生物培养,可直接从基因水平进行分类,是目前环境微生物研究中普遍采用的研究方法之一。本研究采用PCR-DGGE 指纹技术分别在自来水浇灌和污水处理条件下,对5种湿地植物根区土壤细菌群落进行比较分析,探究生活污水对土壤细菌群落及湿地植物根区细菌群落的影响。
1 材料与方法 1.1 试验设置选取银川平原普遍生长的芦苇(Phragmites australis)、水葱(Scirpus validus)、千屈菜(Lythrum salicaria)、扁秆藨草(Scirpus planiculmis)和长苞香蒲(Typha angustata),以这5种湿地植物单独种植构建人工湿地小试系统。模拟试验在塑料桶中进行(桶高34 cm,上口直径34 cm,下口直径27 cm)。
试验地位于银川市植物园实验大棚,自然光照。将野外采集的5种湿地植物带回实验室,挑选生长健壮,大小基本一致的健康植株,单独种植。所用土壤为植物园试验田的沙土,总磷含量0.60 mg·g-1,总氮含量0.43 mg·g-1。每桶种植3株,每种植物种植6桶。用自来水浇灌进行适应性生长,待其生长旺盛,于2012年7月5日进行污水处理试验。将浇灌污水的芦苇、水葱、千屈菜、扁秆藨草和长苞香蒲5种湿地植物设计为处理组,采集土壤样本分别标记为L、S、Q、B和C;将自来水浇灌的5种湿地植物设为对照组ck,采集土壤样本分别标记为ckL、ckS、ckQ、ckB和ckC;将试验所用到的土壤设为背景值1,标记为ck1;将污水浇灌的土壤设为背景值2,标记为ck2。所有对照和处理各设3个重复。试验污水取自宁夏银川市六盘山高级中学南侧校园的排水沟,为教职工生活区排放的生活污水。污水浇灌量为每桶10 L,其水深为12 cm,标记每个水桶的液面作为标准,试验期间通过加自来水补充蒸发和蒸腾所耗的水分,以保持桶中水位。试验于2012年7月5日开始,10月5日结束,为期3个月。不同时期污水污染指标见表 1。
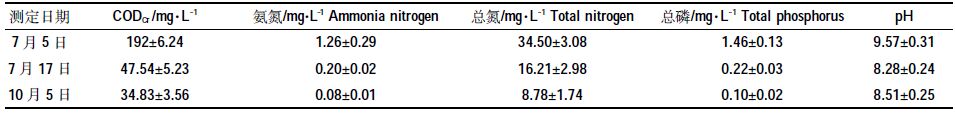
测定试验第12 d(7月17日)各土壤样品中的细菌群落结构(此时污水中总磷、总氮、CODCr等污染指标出现了大幅下降)[10]。于各实验桶中间位置用采泥器旋转采集深度1~10 cm的泥样,各个重复之间充分混匀,取约5 g土样于样品袋中,-80 ℃保存,用于分子生物学分析。
1.3 研究方法 1.3.1 DNA提取及PCR扩增土壤样品中微生物基因组DNA提取采用3s柱离心式环境样品DNA抽提试剂盒提取(上海博彩生物科技有限公司制造)。采用细菌通用引物357F-GC(5′- CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGC-ACGGGGGG CCTACGGGAGGCAGCAG-3′)和5l8R(5′-ATTACCGCGGCTGCTGG-3′)[11]对细菌16s rDNA V3区进行特异性扩增。PCR反应体系为50 μL,其中DNA模板2.5 μL,PCR反应缓冲液25 μL(含TaqDNA聚合酶、Mg2+、dNTP),上游和下游引物各2 μL,加灭菌去离子水18.5 μL至总体积50 μL。采用降落式PCR扩增目的片段,扩增程序有两步。第一步:94 ℃预变性5 min,94 ℃变性45 s,65 ℃退火1 min,72 ℃延伸45 s,共24个循环(每次循环的退火温度比上次循环下降0.5 ℃,24个循环之后退火温度由65 ℃下降到53 ℃);第二步:94 ℃变性45 s,53 ℃退火1 min,72 ℃延伸45 s,再进行11个循环,之后72 ℃延伸5 min。引物357F的5′端加GC夹用来防止DNA片段在进行变性梯度凝胶电泳时过早解链。
1.3.2 DGGE采用变性梯度凝胶电泳仪系统(BIO-RAD DCodeTM Universal Mutation Detection System,USA)对PCR产物进行分析。本试验目的片段为201bp,采用10%的聚丙烯酰胺胶和浓度为40%~70%的变性剂进行电泳。PCR产物上样量为40 μL,在1×TAE缓冲液中60 ℃恒温,160 V电压条件下电泳6 h。电泳后用150 mL 1×TAE加5 μL胶红混合染色30 min,再用Bio-Rad凝胶成像系统拍照。对凝胶上不同泳道的不同和相同条带进行切割(尽量割条带的中间位置),切下条带放入1.5 mL的离心管中,加入PAGE纯化回收试剂盒中的PA溶胶液200 μL,用枪头捣碎,纯化回收。以回收后的DNA为模板,用细菌带夹引物对357F-GC和5l8R按照前述程序进行PCR扩增,扩增产物再用DGGE验证,确定回收条带的正确位置后,再次切胶回收。以第二次回收后的DNA为模板,用不带夹的引物357F和5l8R再进行PCR扩增,扩增产物送北京Invitrogen生物公司测序。
1.3.3 数据分析利用Quantity One凝胶图谱分析软件对DGGE图谱条进行条带配对分析,电泳条带的数量可用来代表细菌的群落丰富度(S);图谱条带可通过计算Shannon-Wiener指数来反映细菌种群结构多样性。Shannon指数用H′来表示,H′的计算是基于DGGE胶条带的位置和条带的强度,而条带的强度则通过条带的峰面积来表示。Shannon指数的公式为:
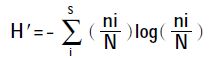
式中:s为泳道的条带总数;ni为泳道第i个条带峰面积;N为一个泳道中所有条带的峰总面积。
测序结果输入NCBI数据库通过BLAST软件与已知序列比对(http://blast.ncbi.nlm.nih.gov/Blast.cgi),获取同源性较高的相关基因序列。利用ClustalX和MEGA5.2软件构建系统进化树。
2 结果与分析 2.1 变性凝胶电泳结果及分析DGGE图谱带数与土样中细菌种群的数量相关,而条带的亮度则在一定程度上反映该种细菌数量的多寡,亮度越高说明该细菌种群数量占比越大。不同处理土壤细菌16s rDNA V3区PCR-DGGE结果如图 1所示。每个样品条带数量和位置差异较大,且优势条带各异,其中:d、k、q带在对照和处理的每个样品中均有出现,且条带亮度基本相同;p、r、s和t带均出现在自来水浇灌的植物样品中,随植物物种不同条带亮度不一;f、g、h、i、j、l、m、n和o带均出现在污水处理样品中,随植物物种不同条带亮度差异较大,其中h条带仅出现在污水处理水葱和千屈菜中,m条带仅出现在污水处理扁秆藨草中,且亮度最高。可以看出污水处理使植物根部土壤中优势细菌群落发生了较大改变。与自来水浇灌相比,污水处理样品细菌条带多而亮,且优势条带明显,说明污水处理过程中植物根部某些细菌大量生长成为优势种。

|
| 图 1 细菌16S rDNA V3 区PCR扩增片段DGGE分析结果 Figure 1 DGGE analysis of 16S rDNA fragments of total bacterial population ck1:试验土壤;ck2:污水处理土壤;L、S、Q、B、C分别为浇灌污水的芦苇、水葱、千屈菜、扁干藨草、长苞香蒲;ckL、ckS、ckQ、ckB、ckC:分别为浇灌自来水的芦苇、水葱、千屈菜、扁干藨草、长苞香蒲;图中所标a~t为切胶回收的条带 ck1:original soil; ck2:sewage soil disposal; Lanes L, S, Q, B, C:Phragmites australis, Scirpus validus, Lythrum salicaria, Scirpus planiculmis, Typha angustata with sewage treatment; Lanes ckL, ckS, ckQ, ckB, ckC:Phragmites australis, Scirpus validus, Lythrum salicaria, Scirpus planiculmis, Typha angustata with tap water treatment; a~t were selected for DNA sequencing and analysis |
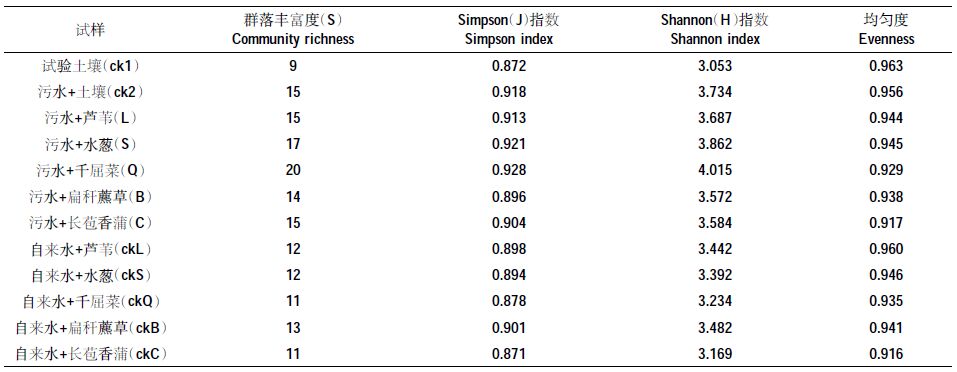
利用Quantity One凝胶图谱分析软件对DGGE图谱进行数字化分析,计算土壤细菌多样性指数如表 2所示。整个DGGE电泳图谱共有29条带,污水处理样品中细菌群落数为15~20个,高于自来水浇灌的11~13个,污水处理植物的多样性指数也均高于自来水浇灌的植物。污水处理的千屈菜根区土壤细菌群落数最高,为20,群落多样性指数也最高,为4.015。
采用非加权组平均法(UPGMA)对变性凝胶图谱中的条带进行聚类分析。从图 2可以看出,自来水浇灌的DGGE指纹图谱最为相似(0.57~0.73,长苞香蒲除外),聚为一类;其次是污水处理植物(芦苇除外)相似性0.50~0.57,聚为一类;试验土壤和污水处理土壤聚为一类,相似性0.58。

|
| 图 2 DGGE电泳图谱聚类分析 Figure 2 Cluster analysis of bacterial communities based upon DGGE |
通过对变性凝胶上优势条带切胶回收测序(图 1中所标a~t),并利用ClustalX和MEGA5.2软件构建系统进化树,进一步分析植物根区细菌类群(试验中共切割送测20条带,其中i、n、p和s未测出结果)。如图 3系统进化树所示,测序得到的16条带中包括5个细菌类群,分别为:变形菌门(Proteobacteria)条带有Band b、Band c和Band e;厚壁菌门(Firmicutes)条带有Band f和Band j;放线菌门(Actinobacteria)条带有Band d;拟杆菌门(Bacteroidetes)条带有Band g、Band h、Band l、Band r、Band t;Uncultured bacterium条带有Band a、Band k、Band o、Band m、Band q。结合图 1和图 3可知试验土壤ck1中的优势细菌有根瘤菌属(Rhizobium)、鞘氨醇单胞菌属(Sphingomonas)和Uncultured bacterium;自来水浇灌植物根区优势细菌有黄杆菌属(Flavobacterium)、节杆菌属(Arthrobacter)和Uncultured bacterium;污水处理土壤ck2的优势细菌有鞘氨醇单胞菌属(Sphingomonas)、根瘤菌属(Rhizobium)、节杆菌属(Arthrobacter)、赤杆菌属(Erythrobacter)和Uncultured bacterium;污水处理植物根区优势细菌有节杆菌属(Arthrobacter)、梭菌属(Clostridium)、硫酸盐还原细菌(Sulfate-reducing bacterium)、牦牛瘤胃菌(Proteiniclasticum)、黄杆菌属(Flavobacterium)和Uncultured bacterium。

|
| 图 3 DGGE图谱中优势条带及其相似菌群系统进化树 Figure 3 Phylogenetic tree based on neighbor-joining analysis of sequences from soil bacteria |
为进一步分析各个样品中细菌群落结构,结合DGGE图谱中条带的峰值及测序结果作图,可得出各植物根区土壤细菌类群结构图(图 4)。植物根区细菌群落随植物物种不同有很大差异。与自来水植物相比,污水处理芦苇根区拟杆菌门细菌占比明显增加,不可培养菌明显减少;水葱根区变形菌门有所增加,放线菌门有所减少;千屈菜根区厚壁菌门和拟杆菌门明显增加,放线菌门有所减少;扁秆藨草根区不可培养菌显著增加,其他各门细菌有所减少;长苞香蒲根区厚壁菌门和放线菌门明显增加,变形菌门有所减少。与试验土壤相比,污水处理土壤和种植植物土壤中拟杆菌门细菌占比增大。

|
| 图 4 植物根区土壤细菌群落结构 Figure 4 The community structure of soil bacteria living in plant root zone soil |
湿地植物污水净化过程中有机氮和磷的去除是一种结合了物理、化学和生物分解的过程,土壤微生物发挥了极其重要的作用[12-13]。本研究采用PCR-DGGE技术检测到5种湿地植物根区细菌共有5个大类,分别为变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)、放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)和不可培养细菌(Uncultured bacterium)。鉴定出细菌8属,分别为赤杆菌属(Erythrobacter)、根瘤菌属(Rhizobium)、鞘氨醇单胞菌属(Sphingomonas)、节杆菌属(Arthrobacter)、黄杆菌属(Flavobacterium)、梭菌属(Clostridium)、硫酸盐还原细菌(Sulfate-reducing bacteria)和牦牛瘤胃菌属(Proteiniclasticum)。
无论是试验土壤、污水处理土壤、自来水浇灌植物根区还是污水处理植物根区,普遍存在变形菌门(Band b)、放线菌门(Band d)、拟杆菌门(Band r)和Uncultured bacterium(Band k、Band q),这几大类群都是报道的湿地植物根区常见的细菌种属,其中不可培养细菌在所有样品中占比较大。本研究中条带d代表放线菌,条带d普遍存在于各植物样本中且条带亮度均一(图 3)。放线菌属于革兰氏阳性菌,在土壤的有机质糖、氨基酸、纤维素和几丁质的降解过程中发挥着重要的作用[14]。变形菌门是细菌中最大的一门,是常见报道的湿地植物根区土壤中微生物群落结构的优势菌群[15]。Ahn 等[16]研究了人工湿地基质沉积物中的微生物群落结构,发现变形菌门占48%~60%,是优势菌群,其次是来自于放线菌门和厚壁菌门。本研究中变形菌门在试验土壤ck1中占比最大,为21%,在其他样品中占比相对较低。在污水处理植物根区检测到大量拟杆菌门中的黄杆菌属(Flavobacterium),其条带有Band g、Band h、Band l。有研究发现黄杆菌属擅长降解高分子物质、蛋白质、脂类纤维素等大分子颗粒有机物,且具有一定的硝化作用和潜在的脱氮能力[17-18],可能与污水中氮的去除有关。此外,在污水处理植物根区还检测到硫酸盐还原细菌(SRB)和鞘氨醇单胞菌。硫酸盐还原细菌是一类以乳酸或丙酮酸等有机物作为电子供体,在厌氧状态下,把硫酸盐、亚硫酸盐、硫代硫酸盐等还原为硫化氢的细菌总称,已广泛运用于废水中硫酸盐的去除[19];而鞘氨醇单胞菌[20]是一类丰富的新型微生物资源,可用于芳香化合物的生物降解,在环境保护及工业生产方面具有巨大的应用潜力。
人工湿地植物根区土壤微生物群落结构和活性会受到诸多因素的影响,例如基质类型、植物种类、有机磷和有机碳的转化、污水的物理化学参数以及污水中的微生物和病原菌等[21]。相关研究表明[22-23]湿地植物根系为土壤中微生物生长提供了重要支撑,其发达的通气组织可将氧气输送至根区土壤中,在植物根区形成好氧区,有利于好氧微生物生长从而明显改变微生物群落结构。Lauber等[24]对种植水生植物的土壤进行微生物群落和理化分析发现,水生植物通过改变土壤pH和土壤理化状况,进而影响微生物群落结构,尤其真菌群落受到的影响作用最为明显。本研究中,与试验土壤ck1相比,自来水浇灌植物和污水处理植物根区的土壤中细菌种群丰富度和多样性指数明显增高(表 2),其中厚壁菌门和拟杆菌门细菌群落增幅最大,变形菌门和放线菌门细菌基本没有变化(图 4),污水处理植物根区细菌种群丰富度和多样性指数高于自来水浇灌植物,各植物之间细菌种群丰富度和多样性指数随植物种类不一,差别不同。与试验土壤ck1相比,污水浇灌土壤ck2中检测到的细菌群落数明显增多,且高于自来水浇灌植物根区细菌群落(表 2)。另外,与污水浇灌土壤ck2相比,污水处理植物根区细菌种群丰富度和多样性指数略有升高,其中污水处理千屈菜根区细菌种群丰富度最高,其次是水葱、芦苇和长苞香蒲扁,秆藨草根区细菌种群丰富度低于污水浇灌土壤ck2。肖烨等[25]对三江平原湿地研究发现总氮与微生物活性指标之间存在极显著正相关关系(P<0.01);Ligi等[26]的研究发现湿地土壤中细菌群落结构与土壤pH及土壤中NH4+-N、NO3--N、Ca和总碳的浓度之间有着复杂的关系;Dong等[17]对人工湿地中微生物的研究结果显示NH4+、TP和4PO3-等营养物质的浓度直接影响细菌多样性和分布情况,其中NH4+对细菌群落的影响最为显著。本研究中用于构建人工湿地小试系统土壤的总磷含量为0.06%,总氮含量为0.043%,氮含量相对较低,且由于植物的吸收作用,自来水浇灌植物根区土壤中营养元素会进一步降低。污水浇灌的土壤,因生活污水富含氮磷,可为细菌提供营养,故其细菌种群数略高于自来水浇灌植物,可见营养元素对土壤中细菌群落的影响作用大于植物本身。这说明污水浇灌土壤ck2和污水处理植物根区的细菌群落与自来水浇灌植物之间发生的差异,与污水中氮、磷、有机物等营养物质有关。
综上所述,PCR-DGGE技术是研究土壤微生物的有效方法,共检测出土壤细菌5个门类的8属,初步探明了生活污水对土壤和湿地植物根区细菌群落造成的影响。浇灌生活污水的土壤ck2和5种植物根区的细菌群落数明显高于自来水浇灌植物ck和试验土壤ck1,说明生活污水明显改变了原有土壤和湿地中的细菌群落结构和多样性。条带测序检测到有与硫分解相关的硫酸盐还原细菌、与芳香化合物降解的鞘氨醇单胞菌、与氮分解相关的黄杆菌属及固氮细菌根瘤菌等,但由于PCR-DGGE技术的缺点[27],试验只测定了土壤中部分优势细菌群落的变化,未检测到在污水净化过程中发挥重要作用的反硝化细菌和氨氧化细菌,不能完全反映土壤微生物的全貌,要全面了解土壤中微生物发生的变化还有待进一步研究。
4 结论(1)浇灌了生活污水的土壤及湿地植物,其细菌种群丰富度和群落多样性指数高于试验土壤和自来水浇灌植物,其中污水处理植物的最高,其次是污水浇灌的土壤和自来水浇灌植物,试验土壤的最低。就污水浇灌土壤,自来水浇灌植物和污水处理植物分别对土壤中细菌群落的影响来看,浇灌污水的土壤和种植湿地植物均明显改变了土壤中的细菌群落结构和多样性,污水对土壤细菌群落多样性的影响作用高于植物。
(2)通过对DGGE条带回收测序检测出土壤细菌5个大类的8属,分别是:变形菌门的根瘤菌属、赤杆菌属、鞘氨醇单胞菌属;厚壁菌门的梭菌属、硫酸盐还原细菌和牦牛瘤胃菌属;放线菌门的节杆菌属;拟杆菌门的黄杆菌属和不可培养细菌,其中不可培养细菌和变形菌门在所有样品中占比均较大。
| [1] | Ye C, Li L, Zhang J, et al. Study on ABR stage-constructed wetland integrated system in treatment of rural sewage[J]. Procedia Environmental Sciences , 2012, 12 (Part A) : 687–692. |
| [2] | Li M, Zhang W, Xia Y, et al. Study on removal efficiencies of pollutant from constructed wetland in aquiculture waste water around Poyang Lake[J]. Procedia Environmental Sciences , 2011, 10 (Part C) : 2444–2448. |
| [3] | Dong H, Qiang Z, Li T, et al. Effect of artificial aeration on the performance of vertical-flow constructed wetland treating heavily polluted river water[J]. Journal of Environmental Sciences , 2012, 24 (4) : 596–601. DOI:10.1016/S1001-0742(11)60804-8 |
| [4] | 杜刚, 黄磊, 高旭, 等. 人工湿地中微生物数量与污染物去除的关系[J]. 湿地科学 , 2013, 11 (1) : 13–20. DU Gang, HUANG Lei, GAO Xu, et al. Number of microbe and relationship between it and removal of pollutants in constructed wetlands[J]. Wetland Science , 2013, 11 (1) : 13–20. |
| [5] | 赵庆节. 种植不同植物的人工湿地土壤微生物群落研究[J]. 上海交通大学学报:农业科学版 , 2011, 29 (3) : 47–52. ZHAO Qing-jie. Study on the soil microbial diversity in the running constructed wetland cultivated with four plants[J]. Journal of Shanghai Jiaotong University(Agricultural Science) , 2011, 29 (3) : 47–52. |
| [6] | 李鑫, 都李萍, 徐婷婷, 等. 植物群落组成对人工湿地微生物群落影响[J]. 生态学杂志 , 2014, 33 (6) : 1508–1514. LI Xin, DU Li-ping, XU Ting-ting, et al. Effects of plant community composition on microbial community in constructed wetlands[J]. Chinese Journal of Ecology , 2014, 33 (6) : 1508–1514. |
| [7] | 王萌, 许新, 陈章和. 人工湿地土壤微生物生物量碳与污水净化效果的关系[J]. 应用与环境生物学报 , 2013, 19 (1) : 113–118. WANG Meng, XU Xin, CHEN Zhang-he. Relatingship of soil microbial biomass carbon and nutrient removal rates in constructed wetlands[J]. Chinese Jounal of Applied and Environmental Biology , 2013, 19 (1) : 113–118. DOI:10.3724/SP.J.1145.2013.00113 |
| [8] | 郭建国, 赵龙浩, 徐丹, 等. 人工湿地处理造纸废水后细菌群落结构变化[J]. 生态学报 , 2014, 34 (8) : 2095–2101. GUO Jian-guo, ZHAO Long-hao, XU Dan, et al. The bacterial community changes after papermaking wastewater treatment with constructed wetlands[J]. Acta Ecologica Sinica , 2014, 34 (8) : 2095–2101. |
| [9] | Torsvik V, ?vre?s L. Microbial diversity and function in soil:From genes to ecosystems[J]. Current Opinion in Microbiology , 2002, 5 (3) : 240–245. DOI:10.1016/S1369-5274(02)00324-7 |
| [10] | 李龙山, 倪细炉, 李志刚, 等. 5种湿地植物生理生长特性变化及其对污水净化效果的研究[J]. 农业环境科学学报 , 2013, 32 (8) : 1625–1632. LI Long-shan, NI Xi-lu, LI Zhi-gang, et al. Growth characteristics and sewage cleaning effect of five wetland plants[J]. Journal of Agro-Environment Science , 2013, 32 (8) : 1625–1632. |
| [11] | Muyzer G, Waal E C, Uitterlinden A G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA[J]. Applied & Environmental Microbiology , 1993, 59 (3) : 695–700. |
| [12] | Tao W, Hall K J, Duff S J. Microbial biomass and heterotrophic production of surface flow mesocosm wetlands treating woodwaste leachate:Responses to hydraulic and organic loading and relations with mass reduction[J]. Ecological Engineering , 2007, 31 (2) : 132–139. DOI:10.1016/j.ecoleng.2007.06.007 |
| [13] | Sundberg C, Sundblad-Tonderski K, Lindgren P-E. Potential nitrification and denitrification and the corresponding composition of the bacterial communities in a compact constructed wetland treating landfill leachates[J]. Water Science and Technology , 2007, 56 (3) : 159–166. DOI:10.2166/wst.2007.524 |
| [14] | Aislabie J M, Chhour K-L, Saul D J, et al. Dominant bacteria in soils of Marble point and Wright valley, Victoria land, Antarctica[J]. Soil Biology and Biochemistry , 2006, 38 (10) : 3041–3056. DOI:10.1016/j.soilbio.2006.02.018 |
| [15] | 关晓燕, 韩家波, 王摆, 等. 辽东湾大凌河口湿地土壤微生物群落分析[J]. 生态环境学报 , 2012, 21 (6) : 1063–1070. GUAN Xiao-yan, HAN Jia-bo, WANG Bai, et al. Analysis of bacterial communities in Liaodong Bay Dalinghe estuarine wetland[J]. Ecology and Environment Sciences , 2012, 21 (6) : 1063–1070. |
| [16] | Ahn C, Gillevet P, Sikaroodi M. Molecular characterization of microbial communities in treatment microcosm wetlands as influenced by macrophytes and phosphorus loading[J]. Ecological Indicators , 2007, 7 (4) : 852–863. DOI:10.1016/j.ecolind.2006.10.004 |
| [17] | Dong X, Reddy G B. Soil bacterial communities in constructed wetlands treated with swine wastewater using PCR-DGGE technique[J]. Bioresource Technology , 2010, 101 (4) : 1175–1182. DOI:10.1016/j.biortech.2009.09.071 |
| [18] | 曾永辉. 典型海洋环境中浮游细菌多样性及环境适应机制的研究[D]. 福建:厦门大学, 2008:1-6. ZENG Yong-hui. Microbial diversity and environmental adaptation mechanisms in typical mariane environments[D]. Fujian:Xiamen University, 2008:1-6. |
| [19] | 曾国驱, 贾晓珊, 郑小红, 等. 硫酸盐还原反应器污泥驯化过程中微生物群落变化分析[J]. 环境科学 , 2014, 35 (11) : 4244–4250. ZENG Guo-qu, JIA Xiao-shan, ZHENG Xiao-hong, et al. Analysis of microbial community variation in the domestication process of sludge in a sulfate-reducing reactor[J]. Environmental Science , 2014, 35 (11) : 4244–4250. |
| [20] | 周丽沙, 李慧, 张颖, 等. 石油污染土壤鞘氨醇单胞菌遗传多样性16S rDNA-PCR-DGGE分析[J]. 土壤学报 , 2011, 48 (4) : 804–812. ZHOU Li-sha, LI Hui, ZHANG Ying, et al. Analysis of Sphingomonas genetic diversity in petroleum-contaminated soils by using PCR-DGGE technique[J]. Acta Pedologica Sinica , 2011, 48 (4) : 804–812. |
| [21] | Truu M, Juhanson J, Truu J. Microbial biomass, activity and community composition in constructed wetlands[J]. Science of the Total Environment , 2009, 407 (13) : 3958–3971. DOI:10.1016/j.scitotenv.2008.11.036 |
| [22] | Dacey J W H. Pressurized ventilation in the yellow waterlily[J]. Ecology , 1981, 62 (5) : 1137–1147. DOI:10.2307/1937277 |
| [23] | Gagnon V, Chazarenc F, Comeau Y, et al. Influence of macrophyte species on microbial density and activity in constructed wetlands[J]. Water Science & Technology A Journal of the International Association on Water Pollution Research , 2007, 56 (3) : 249–254. |
| [24] | Lauber C L, Strickland M S, Bradford M A, et al. The influence of soil properties on the structure of bacterial and fungal communities across land-use types[J]. Soil Biology and Biochemistry , 2008, 40 (9) : 2407–2415. DOI:10.1016/j.soilbio.2008.05.021 |
| [25] | 肖烨, 黄志刚, 武海涛, 等. 三江平原4种典型湿地土壤碳氮分布差异和微生物特征[J]. 应用生态学报 , 2014, 25 (10) : 2847–2854. XIAO Ye, HUANG Zhi-gang, WU Hai-tao, et al. Carbon and nitrogen distributions and microbial characteristics in the soils of four types of wetlands in Sanjiang Plain, Northeast China[J]. Chinese Journal of Applied Ecology , 2014, 25 (10) : 2847–2854. |
| [26] | Ligi T, Oopkaup K, Truu M, et al. Characterization of bacterial communities in soil and sediment of a created riverine wetland complex using high-throughput 16S rRNA amplicon sequencing[J]. Ecological Engineering , 2014, 72 : 56–66. DOI:10.1016/j.ecoleng.2013.09.007 |
| [27] | 夏围围, 贾仲君. 高通量测序和DGGE分析土壤微生物群落的技术评价[J]. 微生物学报 , 2014, 54 (12) : 1489–1499. XIA Wei-wei, JIA Zhong-jun. Comparative analysis of soil microbial communities by pyrosequencing and DGGE[J]. Acta Microbiologica Sinica , 2014, 54 (12) : 1489–1499. |