 2016, Vol. 35
2016, Vol. 35文章信息
- 申连玉, 钱黎慧, 代静玉, 李嘉雨, 周强
- SHEN Lian-yu, QIAN Li-hui, DAI Jing-yu, LI Jia-yu, ZHOU Qiang
- 钙、钠饱和处理的次生硅酸盐矿物对多环芳烃(菲)吸附与光解行为
- Adsorption and photodegradation of phenanthrene on different cation-saturated secondary minerals under UV light
- 农业环境科学学报, 2016, 35(2): 294-304
- Journal of Agro-Environment Science, 2016, 35(2): 294-304
- http://dx.doi.org/10.11654/jaes.2016.02.013
-
文章历史
- 收稿日期: 2015-09-29

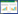




多环芳烃(Polycyclic aromatic hydrocarbon,PAHs)主要产生于机动车尾气排放、化石燃料的燃烧、石油开采、化工原料生产等人类活动过程以及森林火灾或火山爆发等有机污染物的不完全燃烧过程[1, 2]。由于PAHs具有致癌、致畸、致突变等危害以及较低的生物降解性,许多国家都将其列入优先控制检测污染物的黑名单中[3, 4]。
通常,多环芳烃类化合物在水中的溶解性低且辛醇-水分配系数高,进入到环境中的PAHs很容易吸附在土壤或沉积物中进而富集。因此,土壤成为PAHs在自然环境中最重要的储库。在土壤环境中,低分子量PAHs主要通过微生物降解和挥发作用去除,四环或四环以上PAHs水溶性差,很难被微生物降解,但其离域π键可以吸收太阳光的可见(400~700 nm)和紫外(290~400 nm)等部分。因此,光降解作为环境中PAHs转化的一种重要途径而受到广泛重视[5, 6]。
土壤矿物是土壤无机成分的重要部分,在有机污染物的迁移转化降解过程中起到重要作用。土壤矿物主要包括硅酸盐类粘土矿物和金属氧化物矿物。高岭石和蒙脱石是典型的1∶1型和2∶1型层状结构硅酸盐类粘土矿物[7],特殊的晶体结构赋予其特殊的性质,因具有纳-微米级的粒径、较大的比表面积和特殊的纳米层间域而具有良好的吸附和负载性能。蒙脱石由两层硅四氧四面体中间夹一层铝氧八面体构成[8],且结构中的高价元素可被低价元素置换导致结构体电荷失衡,因此层间需要吸附阳离子来平衡电荷[9]。根据层间阳离子种类和含量通常可分为钙基蒙脱石和钠基蒙脱石[10]。根据国内外蒙脱石的分布,目前中国的蒙脱石资源以钙基蒙脱石为主,钠基蒙脱石产量较少。但与钙基蒙脱石相比,钠基蒙脱石具有更强的吸水膨胀性、分散悬浮性、吸附性、粘结性、可塑性、热稳定性、触变性、强度和润滑性、阳离子交换性等特性[11]。因此,钠基蒙脱石较钙基蒙脱石具有更广阔的应用前景和更高的经济价值。另外,水钠锰矿是土壤中最为常见的金属氧化物类矿物,水钠锰矿的比表面积大,在已知氧化锰矿物中,氧化能力最强。同时,水钠锰矿还具有电荷零点(PZC)低,阳离子交换量高等特性,对有机污染物在环境中的氧化还原和吸附解吸过程起着重要的作用。相关研究证实[12, 13],吸附在粘土矿物表面的有机化合物比在其他矿物表面(氢氧化铝、铁氧化物、锰氧化物等)具有更有效的光降解特性。矿物表面负电荷与环境中普遍存在的可交换无机阳离子因相互作用而被中性化,随后改变其表面物理化学性质,同时影响有机化合物与矿物表面的相互作用[14]。
吸附和光化学过程是天然环境中有机污染物迁移转化的重要过程之一,而普遍存在于自然界中的粘土矿物对土壤环境中有机污染物多环芳烃的迁移转化具有非常重要的作用。鉴于此,本文选择两种常见的天然粘土矿物高岭石(1∶1型)、蒙脱石(2∶1型)和实验室合成酸性水钠锰矿及金属离子饱和矿物,研究其对多环芳烃不同吸附特性及紫外光辐射下光降解作用的影响,并通过分析光解产物的种类,探讨可能存在的反应机制,以期为认识粘土矿物和金属氧化矿物在环境中有机污染物迁移转化过程中的作用且为其在污染控制方面的应用提供指导。
1 材料与方法 1.1 矿物的制备与表征为获得高结晶度的层状水钠锰矿,本文采用冯雄汉等[15]的方法实验室合成酸性水钠锰矿。将合成的酸性水钠锰矿粉末压片,采用X射线衍射仪(Dmax-B型,日本理学)进行分析。测定条件为:Cu Kα辐射,管电压30 kV,管电流20 mA,扫描速度为6 °·min-1。
1.2 金属离子饱和矿物与污染矿物的制备金属离子饱和矿物制备:参考并优化王玉军等[16]的方法,各称取2份150 g高岭石(购自国药集团)、蒙脱石(购自阿拉丁,比表面积20~40 m2·g-1)和合成水钠锰矿,分别置于0.2 mol·L-1NaCl和0.2 mol·L-1 CaCl2溶液中饱和过夜,然后将样品进行离心,用去离子水(BECKMAN J2-MC)洗涤若干次至电导率<15 μS·cm-1,且直至上清液中无Cl-(用AgNO3检测)。用冷冻干燥机(Thermo HetoPowerDry LL3000,中国尚马科工贸)将样品冷冻干燥,磨碎,过80目筛备用。
污染矿物制备:在进行污染之前,为保证矿物被均匀污染,所有矿物均玛瑙研钵研磨过80目筛,然后将1000 mg·L-1菲(购自阿拉丁,纯度≥97%)、丙酮(色谱纯)母液稀释后分别滴加至待光照的高岭石、蒙脱石和水钠锰矿及其金属离子饱和矿物,使得其中菲理论含量为60 mg·kg-1,混合均匀,放置通风厨内避光待有机溶剂丙酮挥发完全后,将矿物玛瑙研钵研磨过80目筛,混匀放入-4 ℃冰箱中避光保存待用。
1.3 矿物对PAHs(菲)吸附行为以0.005 mol·L-1CaCl2+100 mg·L-1 NaN3+5 mg·L-1 NaHCO3混合液作为吸附背景溶液,配制浓度分别为0.05、0.1、0.2、0.3、0.4、0.6、0.8、1.0、1.2 mg·L-1的菲标准溶液,其中甲醇的浓度控制在0.2%以下。称取20 mg高岭石、蒙脱石和水钠锰矿至50 mL玻璃离心管,加入25 mL菲标准溶液,将离心管置于恒温(25±1 ℃)培养振荡器(ZWY-240,上海智城)中避光振荡,吸附平衡后,将悬浮液在3000 r·min-1离心10 min,均取10 mL上清液,加10 mL等体积甲醇适当稀释,过0.22 μm微孔滤膜后,用HPLC(2489,美国Waters)测定菲。
1.4 菲光降解实验根据前人研究结果,光穿透土层的平均厚度为0.1~0.5 mm[16],与在预实验获得结果相一致。在2片直径为10 cm圆形石英玻璃片之间夹上外经为10 cm、内径为8 cm的四种不同厚度(0.3、0.5、1.0、3.0 mm)聚氟乙烯垫圈,在中间的(8 cm2×π)石英玻璃面上铺放一定质量的矿物(图 1),在254 nm波长紫外光下进行光照,比较不同厚度聚四氟乙烯垫圈下的光降解效果。结果表明在0.3 mm垫圈下菲去除率最高,因此选择0.3 mm作为后续实验的聚四氟乙烯垫圈厚度。将矿物样品随机摆放至密闭光解反应箱的有效光照区域(光照均匀,光照强度恒定)进行光照。样品与光源的垂直距离为150 mm,以功率为140 W的氙灯作为光源,有效光照区的光强为38.9 mW·cm-2、发射波长为254 nm。将所有含有菲的钙、钠饱和矿物及未加离子矿物样品同时在紫外光辐射下进行光降解80 h,定时取样,从中取1 g移入带旋盖的50 mL玻璃离心管中,加入25 mL色谱级甲醇,放置旋转培养混合器(QB-128,中国QILINBEIER)150 r·min-1恒温避光振荡解吸12 h,3000 r·min-1离心10 min,取上清液过0.22 μm有机滤膜,用HPLC测定菲浓度,紫外检测器波长为245 nm,流动相为甲醇/水(体积比9∶1),流速为1.0 mL·min-1,进样体积为10 μL,数据统计采用Excel 2007等分析软件。

|
| 1.两片石英玻璃片;2.0.3 mm聚四氟乙烯垫圈;3.夹子;4.矿物样品图 1 夹在两层圆形石英玻璃片间矿物样品的放置 Figure 1 Setup of minerals sandwiched between circle quartz glass plates |
对光解24 h和48 h的矿物样品,利用气相色谱-质谱联用仪(320-MS,美国Bruker)和Nexus870型傅里叶变换红外光谱仪对降解过程中多环芳烃中间产物进行鉴定。GC-MS鉴定中间产物时,为了消除样品中杂质的干扰,用二氯甲烷和正己烷(色谱纯)萃取,并按照美国国家环保局方法(method 3630C)对得到的萃取样品进行纯化处理。GC-MS测定条件如下。
色谱条件:用Thermo Trace GC Ultra气相色谱仪。色谱柱DB-5石英毛细柱(30 m×0.25 mm×0.25 μm),进样口温度250 ℃。升温程序:初始温度50 ℃,保持1 min,以25 ℃·min-1升至200 ℃,以8 ℃·min-1升至280 ℃,以1 ℃·min-1升至283 ℃,以2 ℃·min-1升至290 ℃。载气(He)流速1 mL·min-1,进样量1 μL,不分流进样。
质谱条件:电子轰击(EI)离子源,电子能量70 eV,传输线温度260 ℃,离子源温度230 ℃,质量扫描范围(m/z)60~640 amu。
另外,红外光谱数据统计采用Sigmaplot 12.5等进行分析。
2 结果与讨论 2.1 合成水钠锰矿的表征和矿物物性分析合成酸性水钠锰矿和高岭石、蒙脱石及其钙、钠饱和处理矿物的粉晶X射线衍射图谱如图 2所示。图 2a矿物样品的特征峰依次为0.721 3、0.360 3、0.289 3、0.243 2、0.224 6 nm,与JCPDS 01-086-0666相符,无其他矿相衍射峰,表明所合成水钠锰矿为单相水钠锰矿。Na+-高岭石、Ca2+-高岭石(图 2b)的XRD主特征峰从衍射角12.020°和24.821°分别向右偏移至12.523°、25.066°(Na+处理)和12.616°、25.199°(Ca2+处理);Na+-蒙脱石、Ca2+-蒙脱石(图 2c)的XRD主特征峰从衍射角5.700°、17.759°、26.557°、29.340°分别向左偏移至4.347°、17.631°、26.367°、28.411°(Na+处理)和4.673°、17.721°、26.461°、29.001°(Ca2+处理)。对于蒙脱石,根据布拉格定律λ=2×d×sin θ,XRD峰值向左偏移表明衍射角变小,在入射波长λ保持不变的条件下,说明蒙脱石平行原子平面间距d变大。这可能是由于Na+和Ca2+等金属离子掺杂至蒙脱石晶格间导致相应的晶格膨胀而发生衍射峰的偏移。相关文献报道称[17],钙基蒙脱石d001值约为1.55 nm,而钠基蒙脱石约为1.25 nm,与图 2c中未加离子蒙脱石d001(1.549 1 nm)相比,可鉴定本实验所采用的蒙脱石为钙基蒙脱石。高岭石与蒙脱石不尽相同,在钙、钠饱和处理后其矿物主特征峰向右偏移,表明金属离子的掺杂使得高岭石晶格发生收缩现象,导致衍射角变大,因此平行原子平面间距变小。这是由于高岭石晶片与晶片之间形成氢键而结合牢固,晶格距离(0.72 nm)较蒙脱石(0.96~2.14 nm)小、晶层间联结力较蒙脱石强、粘结性和可塑性较蒙脱石弱。在水钠锰矿及其钙、钠饱和处理矿物XRD图谱(图 2d)中,钙、钠饱和处理的水钠锰矿主特征峰未发生明显变化,金属阳离子在水钠锰矿层间主要以三齿共角顶(TCS)的内圈配合物形式存在于八面体空位的上方和下方[18],水钠锰矿如此特殊的金属离子嵌入方式,与硅酸盐类矿物不同,在本实验的饱和浓度及处理条件下,进行金属离子饱和处理后未发生平面层间距d的变化。

|
| 图 2 合成水钠锰矿和未加离子及钙、钠饱和矿物粉晶X 射线衍射图谱 Figure 2 Powder XRD pattern of synthesized δ-MnO2 and cation saturated minerals |
通常,疏水性有机污染物PAHs在极性矿物表面的吸附符合Linear和Freundlich吸附模型[19]。其吸附模型等温式分别为:

式中:Kd为吸附质在液相溶液中和吸附剂之间的分配吸附单位;qe为单位质量吸附剂的吸附量,mg·g-1;Ce为吸附平衡时溶液中的吸附质浓度,mg·L-1;1/n为非线性吸附强度参数;Kf为非线性吸附能力常数,Kf越高吸附能力就越强。
所有样品对菲水溶液的吸附等温线方程及拟合参数见表 1。
 |
比较表 1 Freundlich方程的吸附能力常数Kf可知,钙、钠饱和处理高岭石对菲的吸附能力强于未加金属离子的高岭石;同样的结果发现在蒙脱石与水钠锰矿中。原因是当具有非常强离域π-键的PAHs与缺电子物种阳离子相互作用时形成供电子体系,此时阳离子-π在PAHs吸附到矿物表面过程中起到重要作用且改变矿物表面性质[14, 15, 17, 20, 21]。而比较相同类型硅酸盐类粘土矿物高岭石与蒙脱石对菲的吸附能力时发现(Linear方程的Kd值),未加离子的蒙脱石对菲的吸附能力大于未加离子的高岭石(1.304 2>1.075 3),原因是2∶1型蒙脱石比1∶1高岭石具有更大的比表面积,从而提供更多的吸附位点使有机污染物吸附至其表面;而Na+-高岭石对菲的吸附能力强于Na+-蒙脱石(1.146 2>1.143 5),Ca2+-高岭石对菲的吸附能力弱于Ca2+-蒙脱石(1.058 0<1.185 6)。这可能是不同矿物之间不同吸附特性所引起。高岭石为典型的低电荷或零电荷的层状硅酸盐类粘土矿物,相反,蒙脱石为电荷结构矿物[21, 22, 23],在金属离子饱和过程中参杂至层间域和扩散层的亲水性Na+硬阳离子由于其水化半径大,在蒙脱石表面起阻挡作用,相比在高岭石表面,使得疏水性有机污染物菲不容易在蒙脱石上吸附。相反地,由于只受少量的水化离子的攻击,低电荷矿物高岭石比电荷矿物蒙脱石具有低水化特性,使得其形成不带电的疏水性表面[24]。因此,Na+-高岭石对菲比蒙脱石具有更强的吸附能力和更快的降解速度。这与Jia等[25]报道内容一致。另外,由于矿物表面Ca2+的存在会改变层间水分子的结构,使其矿物表面更容易接收菲等疏水性溶质,而Na+则刚好相反;且二价阳离子比单价阳离子更容易被沉积物吸附,从而减弱沉积物表面的负电性,减小对有机阴离子的排斥作用[26]。因此,Ca2+-蒙脱石比Ca2+-高岭石的吸附能力强,也比Na+-高岭石的吸附能力强。层状硅酸盐类矿物或多或少地表现出一些专性吸附能力,但不及金属氧化矿物[27]。自身具有较强氧化特性的水钠锰矿对菲的吸附量均比高岭石和蒙脱石高。
2.3 矿物-菲体系的光降解矿物-菲体系降解曲线(图 3)显示,光照对菲降解起到不同程度的促进作用。高岭石中紫外辐射与避光条件对菲的去除有明显的区别(图 3a),而蒙脱石中区别并不大(图 3b)。在整个光照80 h内,进行紫外辐射的高岭石及其钙、钠饱和处理的高岭石中菲的去除率均达到90%以上,而避光处理中最高去除率只有39.7%;在相同的紫外辐射时间内,蒙脱石及其钙、钠饱和处理的蒙脱石中菲的去除率最高只有65.7%,而避光处理下最高去除率为26.6%。其原因可能是在经过金属离子饱和处理及冷冻干燥过程中,菲通过Na+-π键吸附在脱水的Na+-蒙脱石表面,在光照过程中,矿物吸收环境中的水分子,蒙脱石表面的Na+通过水合作用很快形成表面水分子膜,导致菲快速脱离矿物表面。因此,水合阳离子可阻碍菲靠近阳离子或矿物表面,最后起到抑制光化学行为作用[28, 29]。另外,与上述吸附行为结合发现,蒙脱石具有层间电荷结构,与高岭石不尽相同,掺杂至层间的金属离子对菲起到了更强的吸附保护作用,使得未被光降解的菲留在其蒙脱石内部孔隙中,不易被解吸出来。

|
| 图 3 菲在不同矿物表面的光降解曲线 Figure 3 Photodegradation curves of phenanthrene on different mineral surfaces |
在光照开始后前30 h内高岭石中菲的降解速率较快,随着光照时间的延长,降解趋于平缓;而蒙脱石中的菲在整个光照时间内随着反应时间的延长降解速率增加缓慢。另外,在光照80 h过程中,菲残留率的对数值ln(Ct /C0)与光照时间t之间呈线性关系,表明菲在矿物表面的光降解为一级反应,而降解速率v等于一级反应动力学常数k与其浓度Ct的乘积,进行拟合得到的动力学方程见图 3。高岭石、Na+-高岭石、Ca2+-高岭石中菲在紫外辐射及其避光处理条件下降解速率分别为:0.054、0.039、0.036 h-1(R2分别为0.930 7、0.831 6、0.958 7)和0.006、0.003、0.003 h-1(R2分别为0.918 2、0.975 3、0.884 2);蒙脱石、Na+-蒙脱石、Ca2+-蒙脱石中分别为:0.014、0.010 4、0.011 h-1(R2分别为0.989 5、0.975 7、0.984 1)和0.001、0.002、0.004 h-1(R2分别为0.965 4、0.948 3、0.872 2)。这说明在紫外辐射条件下菲在钙、钠饱和处理的高岭石和蒙脱石中的光降解速率均低于其未加离子矿物中的菲,其降解速率依次为:高岭石>Na+-高岭石>Ca2+-高岭石、蒙脱石>Ca2+-蒙脱石>Na+-蒙脱石。这可能是由于虽然钙、钠饱和处理使得其对菲吸附能力增强,但外加金属离子后矿物层间或表层存在水化阳离子层,而一般阳离子层可能会阻碍分子扩散迁移甚至阻位,从而影响菲在矿物表面的光降解速率。PAHs分子具有平面结构,当PAHs接近一定几何形状的矿物时趋向与矿物形成对齐的平行结构,因此“阳离子-π距离”为阳离子与PAHs相互作用过程中最重要的影响因素之一[30]。图 2c中钙、钠饱和蒙脱石均比未加离子蒙脱石原子平面间距d变大,当PAHs加入时,具有缺电子和极化性的阳离子吸引芳香环π-供体,促进了电子转移[30]。强阳离子-π键表现更低的稳定性且转化率高[31],因此在紫外辐射条件下菲在Ca2+-蒙脱石去除率大于Na+-蒙脱石。这与Jia等[32]结果一致。而金属饱和蒙脱石对菲较未加离子蒙脱石中去除率低与实验中钙基蒙脱石本身特性也有关。
Gollnick[33]等发现三环或三环以上的碳氢化合物能有效吸收紫外波长而进行自身氧化光降解,因此在本实验中碳氢化合物菲在具有强氧化特性的水钠锰矿表面光降解可由以下两个步骤表示:

首先,菲吸收光子能量后发生电子转移产生电子,随后具有强还原特性的“氧化剂”水钠锰矿接收菲因吸收紫外光而释放的电子被还原成Mn2+[34],进一步促进菲的氧化降解。这表明水钠锰矿具有独特的光化学特性,且反应过程中被还原释放的Mn2+是自然界中锰元素的重要来源[35]。另外,水钠锰矿具有强氧化能力可能与其层状结构内的八面体空穴、层间氢离子较多以及容易接收更多电子有关。从图 3c中可以看出,光照80 h过程中水钠锰矿与硅酸盐类粘土矿物相同,菲在钙、钠饱和处理水钠锰矿中的光降解速率均低于未加金属离子的水钠锰矿中菲的光降解速率。菲光降解速率分别为:0.038、0.036、0.022 h-1(R2分别为0.807 7、0.972 7、0.983 6)和0.026、0.019、0.016 5 h-1(R2分别为0.982 6、0.991 8、0.973 6),其降解速率(紫外光照下)依次为水钠锰矿>Na+-水钠锰矿>Ca2+-水钠锰矿。无光照时,菲在矿物表面的降解是吸附、氧化、催化氧化等共同作用的结果。张嵚等[36]认为由于锰矿物的电荷零点较低,在溶液环境pH值均高于电荷零点时,反应过程中存在的锰矿物表面大量负电荷将阻止有机物及其分解产物在锰矿物表面的吸附。从图 3可以看出,无光照时菲在水钠锰矿中的去除量均较高岭石和蒙脱石中多。考虑到水钠锰矿电荷零点和实际实验环境中pH的关系,在确定吸附作用不是最主要的降解作用的基础上,由于本实验体系反应温度为室温,催化氧化作用也可以排除,因为催化过程一般是锰矿物在高温条件下(高于140 ℃)通过释放晶格氧发生的[37]。因此,氧化作用是避光条件下水钠锰矿对菲降解作用的主要原因,与Yu等[38]研究结果一致。另外,水钠锰矿在无光照条件下对菲表现强降解能力的原因可能与其层状结构内的八面体空穴多、层间氢离子多、活性强等有关[39]。
图 4为相同金属离子饱和处理的不同矿物对菲光降解速率的比较。在紫外辐射条件下,在未加离子及钙、钠饱和处理中菲在高岭石中光降解速率最快,其次为水钠锰矿、蒙脱石;而在避光条件下,水钠锰矿中菲去除速率最快,除了避光Ca2+饱和条件下,在其他所有处理中菲在高岭石避光处理中的降解均比蒙脱石中快。锰氧化物具有良好的吸附性质(吸附能力比高岭石和蒙脱石均强),因此其对菲及降解中间产物可能存在吸附作用,从而直接影响到水钠锰矿光催化能力[36],导致水钠锰矿表面菲进一步的光降解受到抑制。图 5为高岭石、蒙脱石及水钠锰矿分别经过金属离子处理后在紫外辐射和避光条件下对菲光降解速率的比较。除了蒙脱石避光条件,在所有矿物的光照及避光条件下未加离子的矿物中菲去处速率最快。其次在光照条件下高岭石和水钠锰矿中,Na+饱和矿物中菲降解速率大于Ca2+饱和矿物中菲,蒙脱石则相反。

|
| 图 4 相同离子饱和处理的不同矿物对菲去除速率比较 Figure 4 Constantsofreaction rates ofphenanthrene on different minerals saturated with same cation |

|
| 图 5 不同金属离子处理的各矿物对菲去除速率比较 Figure 5 Constants of reaction rates of phenanthrene on a mineral saturated with differentcations |
本实验中,光照0 h时菲浓度与其理论比值即为回收率。污染矿物中菲的理论含量为60 mg·kg-1,而污染过后获得的污染矿物中菲光照0 h时实际菲含量分别为46.48、43.24、29.16 mg·kg-1(高岭石、Na+-高岭石、Ca2+-高岭石);46.18、47.02、47.62 mg·kg-1(蒙脱石、Na+-蒙脱石、Ca2+-蒙脱石);43.88、48.65、27.37 mg·kg-1(水钠锰矿、Na+-水钠锰矿、Ca2+-水钠锰矿)。此结果与上述不同矿物吸附能力差别相符,表 1的Freundlich方程中高岭石及钙、钠饱和处理高岭石的吸附能力常数Kf值分别为0.884 6(高岭石)、0.949 4(Na+-高岭石)、0.902 4(Ca2+-高岭石),钙、钠饱和处理的高岭石Kf值均比未加离子高岭石Kf值高。因此,由于矿物对菲的吸附保护作用,矿物经菲污染后光照前均比理论值60 mg·kg-1小,且未加离子高岭石中菲含量均比钙、钠饱和处理的高岭石中菲含量高;而蒙脱石则相反,在表 1的Linear方程中蒙脱石及其金属离子饱和处理的吸附常数Kd值分别为1.304 2(蒙脱石)、1.143 5(Na+-蒙脱石)、1.185 6(Ca2+-蒙脱石)。未加离子的蒙脱石的Kd值比金属离子饱和处理的高,所以实际光照0 h时菲含量在未加离子蒙脱石中最低。同样的规律可在水钠锰矿的Linear方程Kd值和污染矿物在0 h时菲含量比较中发现。不同菲初始含量也可能是其在矿物表面光降解不同程度影响之一,需要后期实验来证明。
2.4 矿物中菲光降解过程的演变选择硅酸盐粘土矿物蒙脱石和金属氧化物矿物水钠锰矿物光照前后进行FTIR光谱分析,同时对菲在矿物表面光照24 h的二氯甲烷萃取液进行GC-MS分析。图 6a为未加金属离子的水钠锰矿中菲光降解48 h前后的红外光谱图。由下往上矿物样品依次为水钠锰矿光照0 h、水钠锰矿光照48 h、水钠锰矿避光48 h。三种不同样品的FTIR图谱峰型基本保持一致,其中3360 cm-1属于酚羟基或者脂肪族羟基上的O-H伸缩振动吸收,1460、745 cm-1分别为脂肪族C-H的弯曲振动吸收、芳香环上的C=C伸缩振动,1276 cm-1处为羧基上的C-O伸缩振动吸收[40]。从该图中可以看出,光照48 h后水钠锰矿中的1630 cm-1处的吸收较光照前增强,具有不饱和键的PAHs光降解初级产物一般为羟基化合物,进而被氧化成醌或其他小分子有机物,而这些中间产物可能发生了一些缩聚现象形成高分子化合物,从而增强了整体的芳香性。另外还可以发现,避光48 h的水钠锰矿中1276 cm-1处羧基上的C-O伸缩振动吸收、1020 cm-1处糖和脂肪族C-O伸缩振动明显弱于光照48 h的水钠锰矿。

|
| 图 6 不同矿物表面菲光降解的FTIR光谱图 Figure 6 FTIR spectra of different mineral surfaces after phenanthrene photodegradation |
图 6b为蒙脱石及其钙、钠饱和处理的蒙脱石中菲光降解48 h后红外光谱图。由下往上矿物样品依次为Na+-蒙脱石、Ca2+-蒙脱石、蒙脱石。三种不同样品的FTIR图谱峰型基本保持一致,其中3360 cm-1处属于酚羟基或者脂肪族羟基上的O-H伸缩振动吸收,在未加离子的蒙脱石中吸收弱,而在Na+-蒙脱石中响应最强烈,随后是Ca2+-蒙脱石。同样的现象可以在1630 cm-1处吸收峰中发现,振动吸收强度大小依次为蒙脱石
为了探讨菲在矿物表面光降解中间产物的组成,对光照24 h的高岭石、蒙脱石及水钠锰矿用二氯甲烷萃取的上清液利用气相色谱-质谱联用仪进行产物分析,发现三种矿物中菲的光降解中间产物组成大致相同,所以统一得到的结果列于表 2。其中1~6号为酯类、7~10号为酮类、11号为醛类、12~13为羧酸类、14~16号为酚类、17~18号为醇类、19~20号为烷烃类。众所周知,邻苯二甲酸为目前为止菲光降解最主要的光降解产物,且是菲中苯环醌化而形成的9,10-菲醌裂开过程形成的产物[41]。本实验中发现的光解产物1~6号分别为邻苯二甲酸随着时间的增长,其苯环开环而产生的邻苯二甲酸衍生物。而9,10-菲醌物质由于在光照初期产生,迅速进一步开环,形成新的衍生物,因此在光照24 h过程中未能检测出。相关研究证实[5],菲的同分异构体蒽在硅石表面光降解后主要产物也是9,10-蒽醌。再随着时间的推移,邻苯二甲酸的衍生物继续开环氧化进一步生成羧酸类、醇类以及烷烃类等物质。从表 2中产物的保留时间可看出,在光照初期主要产物分别为醛类、酮类、酸类以及酯类等物质,而在后期则是烷烃等脂肪族物质,说明在光照过程中产物不断地被矿化。根据前人的研究结果[39, 42],在光照时间足够长时,所有这些中间产物均将可能全部被矿化成水和CO2等小分子物质,而本实验中的中间产物只代表整个光解过程中所有产物的一部分,因为有些产物具有很短的“寿命”未能在本检测条件下被检测出。

图 7为根据实验中所检测到的菲紫外光降解的中间产物而推测的降解途径。反应过程中生成的R·或·OH、·OOH等自由基与降解过程中产生的菲中间产物相互反应、氧化,最终转化为烷烃或脂肪酸与酯,并且在光照时间足够长时,可能均将彻底被矿化成水和CO2等小分子物质[42]。

|
| 图 7 菲在矿物表面光解途径 Figure 7 Pathways of phenanthrene photodegradation on mineral surfaces |
对于高岭石、蒙脱石、水钠锰矿,金属离子饱和处理矿物对菲的吸附能力较未加离子矿物对菲的吸附能力强,相同类型硅酸盐类粘土矿物中,Na+饱和的1∶1型高岭石吸附能力强于Na+饱和2∶1型蒙脱石,Ca2+饱和矿物则相反,而自身具有强氧化特性的水钠锰矿对菲的吸附能力比高岭石和蒙脱石强。在紫外辐射下,未加离子的高岭石、蒙脱石、水钠锰矿中菲光降解速率均大于金属离子饱和矿物表面菲光降解速率。在所有未加离子和金属离子饱和处理的矿物中,高岭石表面菲光降解速率最快,其次为水钠锰矿和蒙脱石。紫外辐射后矿物表面芳香性增强;同时,GC-MS检测不同矿物表面菲光降解产物种类大致相同,主要为邻苯二甲酸衍生物等酯类以及其继续开环氧化生成的羧酸类、醇类以及烷烃类等物质,且在辐射条件足够长时,这些有机物均会彻底被矿化成水和CO2等小分子物质。
| [1] | Fan S M. Photochemical and biochemical controls on reactive oxygen and iron speciation in the pelagic surface ocean[J]. Marine Chemistry, 2008, 109(1):152-164. |
| [2] | Peng C, Chen W, Liao X, et al. Polycyclic aromatic hydrocarbons in urban soils of Beijing:Status, sources, distribution and potential risk[J]. Environmental Polluttion, 2011, 159(3):802-808. |
| [3] | Page S E, Arnold W A, Kristopher M N. Assessing the contribution of free hydroxyl radical in organic matter-sensitized photohydroxylation reactions.[J]. Environmental Science & Technology, 2011, 45(7):2818-2825. |
| [4] | Tobiszewski M, Namieśnik J. PAH diagnostic ratios for the identification of pollution emission sources[J]. Environmental Pollution, 2012, 162(5):110-119. |
| [5] | Librando V, Bracchitta G, Guidi G D, et al. Photodegradation of anthracene and benzo[a] anthracene in polar and apolar media:New pathways of photodegradation[J]. Polycyclic Aromatic Compounds, 2014, 34(3):263-279. |
| [6] | Ma J, Liu Y, Ma Q, et al. Heterogeneous photochemical reaction of ozone with anthracene adsorbed on mineral dust[J]. Atmospheric Environment, 2013, 72(6):165-170. |
| [7] | 吴平霄. 粘土矿物材料与环境修复[M]. 北京:化学工业出版社, 2004:264-280. WU Ping-xiao. Clay mineral materials and environmental restoration[M]. Beijing:Chemical Industry Press, 2004:264-280. |
| [8] | Valandro S R, Poli A L, Neumann M G, et al. Organomontmorillonite/poly(methyl methacrylate) nanocomposites prepared by in situ photopolymerization:Effect of the organoclay on the photooxidative degradation[J]. Applied Clay Science, 2013, 85(6):19-24. |
| [9] | 邱俊, 崔学奇, 吕宪俊, 等. 对蒙脱石层电荷的测定[J]. 矿业快报, 2005, 21(6):6-9. QIU Jun, CUI Xue-qi, LÜ Xian-jun, et al. Measurement of montmorillonite layer charge[J]. Express Information of Mining Industry, 2005, 21(6):6-9. |
| [10] | 苗春省. X射线衍射快速划分膨润土类型的方法[J]. 矿物学报, 1984, 1:88-89. MIAO Chun-sheng. A rapid approach to classifying the types of expandable soils by X-ray diffraction[J]. Acta Mineralogica Sinica, 1984, 1:88-89. |
| [11] | 姜桂兰. 膨润土加工与应用[M]. 北京:化学工业出版社, 2005. JIANG Gui-lan. Bentonite processing and application[J]. Beijing:Chemical Industry Press, 2005. |
| [12] | Li K, Ferry J L. Effect of salinity on the photolysis of chrysene adsorbed to a smectiteclay[J]. Environmental Science & Technology, 2003, 37(21):4894-4900. |
| [13] | Mi-Youn A, Filley T R, Jafvert C T, et al. Photodegradation of decabromodiphenyl ether adsorbed onto clay minerals, metal oxides, and sediment[J]. Environmental Science & Technology, 2006, 40(1):215-220. |
| [14] | Zhang W, Zhuang L, Yuan Y, et al. Enhancement of phenanthrene adsorption on a clayey soil and clay minerals by coexisting lead or cadmium[J]. Chemosphere, 2011, 83(3):302-310. |
| [15] | 冯雄汉, 翟丽梅, 谭文峰, 等. 几种氧化锰矿物的合成及其对重金属的吸附和氧化特性[J]. 岩石矿物学杂志, 2005, 24(6):531-538. FENG Xiong-han, ZHAI Li-mei, TAN Wen-feng, et al. The syntheses of several Mn oxide minerals and their adsorption and redox characteristics for heavy metals[J]. Acta Petrologicaet Mineralogica, 2005, 24(6):531-538. |
| [16] | 王玉军, 周东美, 孙瑞娟, 等. 除草剂草甘膦在几种土壤和矿物上的吸附研究[J]. 土壤学报, 2006, 43(5):780-785. WANG Yu-jun, ZHOU Dong-mei, SUN Rui-juan, et al. Adsorption of glyphosate on soils and minerals[J]. Acta Pedologica Sinica, 2006, 43(5):780-785. |
| [17] | 侯梅芳, 马北雁, 万洪富, 等. 我国各地膨润土的矿物学性质[J]. 岩矿测试, 2002, 21(3):190-194. HOU Mei-fang, MA Bei-yan, WAN Hong-fu, et al. Mineralogical properties of various bentonites in China[J]. Rock and Meneral Analysis, 2002, 21(3):190-194. |
| [18] | 殷辉, 冯雄汉, 邱国红, 等. 掺钴水钠锰矿对铅的吸附及对砷的氧化[J]. 环境科学, 2011, 32(7):2092-2101. YIN Hui, FENG Xiong-han, QIU Guo-hong, et al. Lead adsorption and arsenite oxidation by cobalt doped birnessite[J]. Environmental Science, 2011, 32(7):2092-2101. |
| [19] | 刘晓华, 吴宏海, 管玉峰, 等. 蒙脱石与滑石矿物对菲吸附-解吸行为的对比研究[J]. 矿物岩石, 2011, 31(3):6-10. LIU Xiao-hua, WU Hong-hai, GUAN Yu-feng, et al. Comparison of sorption-desorption behaviors of both montmorillonite and talc minerals for phenanthrene[J]. Journal of Mineral and Petrology, 2011, 31(3):6-10. |
| [20] | Hebert V R, Miller G C. Depth dependence of direct and indirect photolysis on soil surfaces[J]. Journal of Agricultural and Food Chemistry, 1990, 38(3):913-918. |
| [21] | Goss K U, Schwarzenbach R P. Linear free energy relationships used to evaluate equilibrium partitioning of organic compounds[J]. Environmental Science & Technology, 2000, 35(1):1-9. |
| [22] | Ding S, Zhang L, Ren X, et al. The characteristics of mechanical grinding on kaolinite structure and thermal behavior[J]. Energy Procedia, 2012, 16(Part B):1237-1240. |
| [23] | Ramadan A R, Esawi A M K, Gawad A A. Effect of ball milling on the structure of Na+-montmorillonite and organo-montmorillonite(Cloisite 30B)[J]. Applied Clay Science, 2010, 47(3):196-202. |
| [24] | Laird D A. Layer charge influences on the hydration of expandable 2:1 phyllosilicates[J]. Clays & Clay Minerals, 1999, 47(5):630-636. |
| [25] | Jia H, Zhao J, Fan X, et al. Photodegradation of phenanthrene on cation-modified clays under visible light[J]. Applied Catalysis B Environmental, 2012, 123-124(12):43-51. |
| [26] | 陶庆会, 汤鸿霄. 阿特拉津在天然水体沉积物中的吸附行为[J]. 环境化学, 2004, 23(2):145-151. TAO Qing-hui, TANG Hong-xiao. Study on the sorption behavior of atrazine by natural aquatic sediments[J]. Environmental Chemistry, 2004, 23(2):145-151. |
| [27] | Chien S W C, Chang C H, Chen S H, et al. Oxidative degradation of pyrene in contaminated soils by δ-MnO2 with or without sunlight irradiation[J]. Science of the Total Environment, 2011, 409(19):4078-4086. |
| [28] | Zhu D, Herbert B E, Schlautman M A, et al. Cation-π bonding:A new perspective on the sorption of polycyclic aromatic hydrocarbons to mineral surfaces[J]. Journal of Environmental Quality, 2004, 33(4):1332-1330. |
| [29] | Tamara P, Yona C, Rotem N, et al. Interactions of hydrophobic fractions of dissolved organic matter with Fe3+-and Cu2+-montmorillonite[J]. Environmental Science & Technology, 2008, 42(13):4797-4803. |
| [30] | Si Y, Wang S, Zhou D, et al. Adsorption and photo-reactivity of bensulfuron-methyl on homoionic clays[J]. Clays & Clay Minerals, 2004, 52(6):742-748. |
| [31] | Gunilla S, Ulla S, Wit C A D, et al. Photolytic debromination of decabromodiphenyl ether(BDE 209)[J]. Environmental Science & Technology, 2004, 38(1):127-132. |
| [32] | Jia H, Li L, Chen H, et al. Exchangeable cations-mediated photodegradation of polycyclic aromatic hydrocarbons(PAHs) on smectite surface under visible light[J]. Journal of Hazardous Materials, 2015, 287C:16-23. |
| [33] | Gollnick K, Schenck G O. Chapter 10-oxygen as a dienophile[J]. Organic Chemistry, 1967, 8:255-344. |
| [34] | Rennert T, Pohlmeier A, Mansfeldt T. Oxidation of ferrocyanide by birnessite[J]. Environmental Science & Technology, 2005, 39(3):821-825. |
| [35] | Kwon K D, Refson K, Sposito G. On the role of Mn(IV) vacancies in the photoreductive dissolution of hexagonal birnessite[J]. Geochimica Et Cosmochimica Acta, 2009, 73(14):4142-4150. |
| [36] | 张嵚, 冯雄汉, 邱国红, 等. 不同氧化锰矿物对光催化降解苯酚的影响[J]. 矿物学报, 2011, 3(2):263-273. ZHANG Qin, FENG Xiong-han, QIU Guo-hong, et al. Photocatalytic degradation of phenol with manganese oxides:The effects of crystal structure and reaction mechanisms[J]. Acta Mineralogica Sinica, 2011, 3(2):263-273. |
| [37] | Luo J, Zhang Q H, Garcia-Martinez J, et al. Adsorptive and acidic properties, reversible lattice oxygen evolution, and catalytic mechanism of cryptomelane-type manganese oxides as oxidation catalysts[J]. J Am Chem Soc, 2008, 130(10):3198-3207. |
| [38] | Yu J, Savage P E. Catalyst activity, stability, and transformations during oxidation in supercritical water[J]. Applied Catalysis B Environmental, 2001, 31(2):123-132. |
| [39] | Zhao W, Feng X H, Tan W F, et al. Relation of lead adsorption on birnessites with different average oxidation states of manganese and release of Mn2+/H+/K+[J]. Journal of Environmental Sciences, 2009, 21(4):520-526. |
| [40] | Jacquot F, Guiliano M, Doumenq P, et al. In vitro photooxidation of crude oil maltenic fractions:Evolution of fossil biomarkers and polycyclic aromatic hydrocarbons[J]. Chemosphere, 1996, 33(4):671-681. |
| [41] | Zhang Y, Wong J W C, Liu P, et al. Heterogeneous photocatalytic degradation of phenanthrene in surfactant solution containing TiO2 particles.[J]. Journal of Hazardous Materials, 2011, 191(1-3):136-143. |
| [42] | Kou J, Yong G, Gao J, et al. Photocatalytic degradation of polycyclic aromatic hydrocarbons in GaN:ZnO solid solution-assisted process:Direct hole oxidation mechanism[J]. Journal of Molecular Catalysis A Chemical, 2010, 325(1):48-54. |