 2017, Vol. 36
2017, Vol. 36文章信息
- 王琳, 王睿, 郑循华, 张丽娟
- WANG Lin, WANG Rui, ZHENG Xun-hua, ZHANG Li-juan
- 气相转化-化学发光法测定土壤中的亚硝态氮
- A gas phase conversion-chemiluminescence method for detecting nitrite concentrations in soils
- 农业环境科学学报, 2017, 36(9): 1916-1924
- Journal of Agro-Environment Science, 2017, 36(9): 1916-1924
- http://dx.doi.org/10.11654/jaes.2017-0238
文章历史
- 收稿日期: 2017-02-28

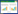




2. 中国科学院大气物理研究所大气边界层物理和大气化学国家重点实验室, 北京 100029;
3. 中国科学院大学地球科学学院, 北京 200049
2. State Key Laboratory of Atmospheric Boundary Layer Physics and Atmospheric Chemistry, Institute of Atmospheric Physics, Chinese Academy of Sciences, Beijing 100029, China;
3. College of Earth Sciences, University of Chinese Academy of Sciences, Beijing 200049, China
亚硝态氮(NO2-)在陆地生态系统氮循环过程中具有重要作用,参与土壤的多个氮素转化过程,如硝化、反硝化、硝化细菌反硝化、厌氧氨氧化及硝酸盐异化还原为铵等过程[1],可生成一氧化氮(NO)和氧化亚氮(N2O)等含氮气体,间接影响气候及环境。其中,NO是大气中的高活性组分,会造成空气质量恶化,还会导致酸雨的形成和对流层臭氧(O3)的产生[2-3];N2O是一种重要的温室气体,也是破坏平流层O3的首要人为源大气成分[4]。土壤NO2-还是大气中气态亚硝酸(HONO)的重要来源[5],HONO是城市污染的一种典型代表物,大气中氢氧自由基主要来源于HONO光解,土壤NO2-的含量与转化对于大气化学及气溶胶动力学有着重要的影响[6]。此外,NO2-会通过植物进入人体内,一方面会降低血液的载氧能力,导致高铁血红蛋白症[7];另一方面可与次级胺结合,形成亚硝胺,诱发癌变[8-9]。因此,准确地测定土壤中NO2-的含量对于氮素循环、气候环境变化研究及人类健康评估具有重要的影响。
测定NO2-的方法有比色法[10-11]、荧光光度法[12-13]、化学发光法[14-15]、气相色谱与质谱联用法[16]、离子色谱法[17-18]和流动分析法[19-20]等。比色法最为常用,测定水质中NO2-含量的国标方法就是比色法[21],其测定原理是NO2-在磷酸介质(pH=1.8)中与4-氨基苯磺酰胺反应生成重氮盐,再与N-(1-萘基)-乙二胺二盐酸盐偶联生成红色染料,在540 nm波长处测定其吸光度,当NO2-含量小于200 μg·L-1(以纯N计,下同)时,吸光度与NO2-含量成正比。有研究者采用将NO2-还原转化为NO气体与化学发光法检测NO相结合的方法(气相转化-化学发光法,简称NC法)测定水体及血浆中微量的NO2-。该方法的原理是在酸性条件下,NO2-被还原为NO,产生的NO与O3反应,生成激发态的二氧化氮(NO2*),其在返回基态时放出的能量与NO2-浓度成正比,由此可对NO2-进行定量(见反应式1、2,其中hv表示释放出的能量)。1980年Cox[22]首次用该方法测定自然水体中的NO2-,还原剂为碘离子(I-);之后有研究者利用钒(V3+)作为还原剂测定海水中的NO2-[23-24];还有研究者利用抗坏血酸(Vc)作为还原剂,检测血浆中的NO2-浓度[25]。

NO2-是活性很强的离子,极易发生反应而转化成其他的氮素形态,因而土壤或浸提液样品中的NO2-不易保存,大量样品难以实现在线测定。研究中往往通过加入稳定剂抑制微生物活性的方法保存NO2-,但稳定剂大多有毒性(如氯仿、氯化汞等)[26],为此需要寻找一个能够直接、在线测定土壤NO2-含量的简易而安全的方法。Nagababu等[25]采用NC法在线测定血浆中痕量的NO2-,操作相对简单并具有较高的准确度(97%)和低的检测限(0.2~0.9 μg·L-1)。但目前仅有少数研究者采用NC法测定土壤浸提液中的NO2-,如Amini等[27]。NC法能否成为一种在线测定土壤中NO2-含量的方法是本研究关注的科学问题。本文参照Nagababu等[28]提供的测定血浆中NO2-的NC法(利用冰醋酸提供酸性条件,抗坏血酸作为还原剂),测定三种不同酸碱度土壤的NO2-浓度,并与比色法的测定结果进行比较,评估该方法对于测定土壤NO2-含量的分析精度及适用性。
1 材料与方法 1.1 试验设计使用亚硝酸钠(NaNO2)配制标准工作液,选取中间浓度的标准溶液采用NC法重复测定,通过变异系数计算方法的精度(浓度与其2倍变异系数的乘积);用浓度稀释法在3倍信噪比时,测得NO2-的浓度,求得检出限;采用NC法测定标准工作液中NO2-的浓度,确定该方法适宜的检测范围。
选择三种不同酸碱度的土壤,按5:1的水土比,分别采用去离子水和含有50 μg·L-1的NaNO2溶液浸提,所得浸提液分别称为待测液A和待测液B;将待测液B与50 μg·L-1的NaNO2溶液等体积混合,得到待测液C,以计算方法的回收率。每个土壤设置3次重复,以试验用水作为空白对照,待测液A、B和C中的NO2-浓度同时采用比色法和NC法测定,并分别计算回收率,评估NC法测定土壤NO2-的准确度和适用性。
1.2 供试土壤及浸提液制备供试土壤分别为取自湖南省长沙市金井镇小流域(113°20′E,28°33′N)双季稻田的酸性(pH 5.4)砂质壤土(HN)、取自湖北省十堰市西沟村(110°37′E,32°38′N)单季稻田的酸性(pH 6.2)砂质壤土(HB)和取自山西省永济市董村农场(34°56′N,110°43′E)的典型冬小麦-夏玉米轮作农田的碱性(pH 8.7)壤质粘土(SX)。土壤的基本理化性质见表 1,详细的试验点概况及土壤性质数据请参见文献[29-31]。采回的鲜土经风干、过筛(2 mm)、除杂、混匀后,置于室内干燥保存。
称取风干土壤样品20.00 g置于三角瓶中,加入100 mL去离子水或50 μg·L-1的NaNO2溶液,振荡30 min,离心(8000 r·min-1)5 min,过滤(0.45 μm聚醚砜膜),浸提液当天测定。
1.3 试验方法 1.3.1 NC法测定NO2-浓度(1)仪器和试剂
氮氧化物分析仪(42i NO-NO2-NOx,热电环境仪器公司,中国上海),当平均读数时间为60 s时,仪器的检出限为0.40 nmol·mol-1。
NaNO2标准贮备液1(100 mg·L-1):称取0.492 9 g的NaNO2试剂溶解于去离子水中,定容至1 L,4 ℃冰箱内可保存一周。
NaNO2标准贮备液2(1 mg·L-1):吸取1 mL的贮备液1,用去离子水定容至100 mL,配制仅供当天使用。
NaNO2标准工作液:吸取0.1、0.5、1.0、1.5、2.0、2.5、5.0、10.0、15.0、25.0 mL的贮备液2于50 mL容量瓶中,用去离子水定容,获得浓度为2、10、20、30、40、50、100、200、300、500 μg·L-1的标准工作液。
50%的冰醋酸溶液:将冰醋酸和去离子水等体积混合。
抗坏血酸溶液(0.05 mol·L-1):称取0.88 g抗坏血酸溶解于100 mL去离子水中,配制仅供当天使用。
氢氧化钠(NaOH)溶液(2 mol·L-1):称取80 g NaOH溶于1 L去离子水中。
(2)试验装置
装置由气源、反应部分、检测和数据采集三部分组成(图 1)。气源为高纯氮气(N2,99.999%),用质量流量控制器调节流速为500 mL·min-1,以满足氮氧化物分析仪的进气要求。反应部分包括两个广口瓶,一个装有20 mL 50%的冰醋酸溶液和3 mL 0.05 mol·L-1抗坏血酸溶液(反应瓶),另一个装有20 mL 2 mol·L-1的NaOH溶液(除酸瓶),分别于瓶口涂上凡士林并用橡胶塞塞紧,在反应瓶的胶塞上固定好长型针头用于进样。检测和数据采集部分包括氮氧化物分析仪、电脑和自动数据采集软件,该软件以1 Hz频率将氮氧化物分析仪的NO浓度信号值记录到电脑中。

|
| 图 1 气相转化-化学发光法(NC法)测定NO2-的试验装置图 Figure 1 The device of chemiluminescence method for determination of NO2- concentration |
由于大气中常含有高浓度的NO,尤其是雾霾天气时大气本底的NO浓度能高达100 nmol·mol-1以上,装置的气密性直接关系到NO2-测定的准确性,因此在试验前严格检查装置的气密性,保证其在试验过程中不受外界NO的干扰。首先检测反应部分的气密性,在两个广口瓶瓶口涂上凡士林,塞紧胶塞,堵住胶塞一端的管路,另一端通入0.2 MPa的N2,将广口瓶浸入水中,观察瓶口是否有气泡产生,若没有表明广口瓶气密性良好;然后检测整个管路的气密性,将肥皂水泡沫涂于管路连接处,通入0.2 MPa的N2观察是否有气泡产生,若没有表明整个管路气密性良好。
(3)试验方法
将高纯N2、装有冰醋酸和抗坏血酸的反应瓶及装有NaOH的除酸瓶依次连接,打开气源使N2流经反应部分,约2 h后,连入氮氧化物分析仪,待反应瓶背景NO浓度降低至5 nmol·mol-1以下并稳定,于室温条件(约25 ℃)下测定标准工作液和待测液中的NO2-浓度。
用注射器吸取1 mL标准工作液/待测液,通过长型针头加入到反应瓶底部溶液中。反应瓶中产生的NO随N2进入氮氧化物分析仪,NO浓度的信号值变化通过数据采集软件实时采集并保存到电脑中。待NO浓度降低至5 nmol·mol-1以下并稳定,可再次进样测定,每次进样前用待测液润洗长型针头。每个浓度的工作液重复测定3次,每测定约含500 ng NO2--N样品更换一次反应瓶。
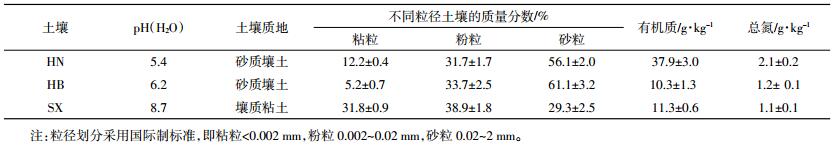
测定结束后,对标准工作液和待测液的NO浓度值的峰面积进行积分,通过标准工作液NO2-浓度与其对应峰面积的线性回归方程(公式1),将待测样品的峰面积带入公式1可计算其NO2-的浓度。
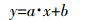 (1)
(1) 式中:y为NO2-标准工作液浓度,μg·L-1;x为NO2-标准工作液相应的NO信号值峰面积;a、b为拟合参数。
1.3.2 比色法测定NO2-浓度[21](1)仪器与试剂
分光光度计(UNICO UV-2802,尤尼柯仪器有限公司,中国上海)
NaNO2标准贮备液1、2的配制方法同1.3.1。
NaNO2标准工作液:吸取0.5、2.5、5.0、7.5、10.0 mL的贮备液2于50 mL容量瓶中,用去离子水定容,获得浓度为10、50、100、150、200 μg·L-1的标准工作液。
显色剂:500 mL烧杯内置入250 mL去离子水和50 mL磷酸(15 mol·L-1),加入20.0 g 4-氨基苯磺酰胺,再将1.00 g N-(1-萘基)-乙二胺二盐酸盐溶于上述溶液中,转移至500 mL棕色容量瓶中,用去离子水稀释至标线,摇匀,置于4 ℃冰箱保存。
(2)试验方法
标准工作液测定:向50 mL的标准工作液中加入1.0 mL显色剂,密塞,摇匀,静置。20 min后、2 h以内,在540 nm波长处测定吸光度。
样品测定:将选定体积的样品移至50 mL比色管中,用水稀释至刻度线,之后的操作同标准工作液的测定方法。
测定结束后,进行标准工作液的NO2-浓度与相应吸光度的线性回归。样品吸光度通过空白参比校正后,根据回归方程计算NO2-浓度,计算方法同1.3.1。
(3)回收率的计算
大多研究者通过加入待测物的实测量除以加入量来计算方法回收率[24, 32]。土壤待测液B与A中NO2-浓度差减的理论值应该为浸提待测液B时所用的NaNO2浓度(50 μg·L-1),回收率(RB-A)如公式2所示。但土壤中加入的50 μg·L-1 NO2-可能被微生物消耗而导致回收率降低。因此,本研究通过待测液C和B的差值与NO2-加入量的比值计算回收率(RC-B),如公式3所示。
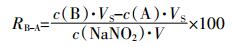 (2)
(2) 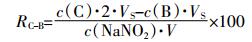 (3)
(3) 式中:R为方法回收率,%;c(A)、c(B)和c(C)分别为待测液A、B和C中NO2-的浓度,μg·L-1;c(NaNO2)为加入的NaNO2标准溶液的浓度,50 μg·L-1;VS为待测液体积,L;V为加入的50 μg·L-1 NaNO2标准溶液的体积,L。
1.4 统计检验采用Microsoft Office Excel 2010进行数据的积分处理,采用SPSS 16.0的独立样本t检验进行P<0.05水平的显著性检验,采用Microsoft Office Excel 2010绘图。
2 结果与分析 2.1 NC法冰醋酸浓度的选择Nagababu等[28]采用NC法测定血浆中NO2-浓度时,选择7 mL纯冰醋酸和1 mL 0.05 mol·L-1抗坏血酸作为反应剂,且每测定4个3 μg·L-1左右的NO2-样品(总含氮量1.2 ng NO2--N)更换一次反应瓶。考虑到土壤的NO2-浓度高于血浆,如曹娜等[33]观测到厌氧条件下土壤中NO2-浓度可达200 μg·L-1,本研究将反应瓶中的反应剂体积增加2倍,改为20 mL 99.5%的冰醋酸与3 mL 0.05 mol·L-1的抗坏血酸混合,既保证对高浓度NO2-的分析能力,又不用频繁更换反应瓶。但是当反应罐中只装有20 mL 99.5%冰醋酸,高纯N2通过反应罐进入氮氧化物分析仪时,可观测到很高的NO浓度背景,最高可达1000 nmol·mol-1,随后NO浓度逐渐降低,待背景NO信号值降至5 nmol·mol-1且稳定则至少需要5 h;在此期间若晃动反应瓶或加入抗坏血酸溶液均会导致NO信号值增大(至80 nmol·mol-1左右),待信号重新降至5 nmol·mol-1且稳定又需要2 h的时间。这远高于Nagababu等[28]给出的测样之前的稳定时间(0.5~1 h),如此长的稳定时间可能是反应剂体积增加造成的。
考虑到冰醋酸在NO2-的还原反应中只起到提供酸性环境的作用,本研究将冰醋酸浓度稀释至50%,在不改变抗坏血酸浓度的条件下与其混合,即采用20 mL 50%的冰醋酸和3 mL 0.05 mol·L-1抗坏血酸。结果显示反应瓶背景的NO浓度能够在2 h内降低至5 nmol·mol-1以下并稳定,而且在50%的冰醋酸条件下重复测定10次50 μg·L-1 NO2-的稳定性很好(参见下节及图 2)。

|
| 图 2 重复测定10次50 μg· L-1 NaNO2标准溶液的NO浓度图 Figure 2 The chemiluminescence concentration of NO by ten times repeated measurement of 50 μg·L-1 in NaNO2 standard solution |
采用NC法重复测定10次50 μg·L-1 NO2-标准工作液,其对应的NO信号值峰虽然有降低的趋势(图 2),但是峰面积稳定,平均值为6 573.1±76.7(n=10),CV为1.2%,95%置信区间内的精度为1.2 μg·L-1,即对于土壤样品的检测精度为6 μg·kg-1(按水土比5:1计)。该结果表明,50%的冰醋酸提供的酸性环境可以满足对50 μg·L-1 NO2-的10次测定,且每测定500 ng NO2--N需更换一次反应瓶。
用浓度稀释法在3倍背景浓度的信噪比时,测得NO2-的检出限为2 μg·L-1,相当于2 ng的NO2--N(进样量为1 mL)。采用NC法测定NO2-标准工作液系列,浓度范围为2~500 μg·L-1,其对应的氮氧化物分析仪NO浓度信号变化如图 3a所示。每个浓度的标准工作液重复测定3次的CV值介于0.3%~5.2%之间,平均为2.5%。NO2-的浓度与NO浓度值峰面积显著正相关(P < 0.001,图 3b)。

|
| 图 3 NO2-标准工作液的NO浓度信号图(a)和标准曲线(b) Figure 3 Chemiluminescence NO concentration of nitrite standard solution (a)and standard curve (b) |
同时采用NC法和比色法测定3种不同类型土壤NO2-浓度的结果如图 4所示。两方法测定HB和HN土壤的NO2-浓度差异不显著,但对于SX土壤,NC法的测定结果较比色法显著偏高10.1%~21.3%(P<0.05)。HB和HN土壤本底(待测液A)的NO2-浓度较低,为8.0~8.5 μg·L-1(比色法)和6.2~7.9 μg·L-1(NC法);SX土壤本底的NO2-浓度为34.6 μg·L-1(比色法)和42.9 μg·L-1(NC法),较HB和HN土壤高。

|
| 不同小写字母表示两种方法间存在显著差异(P < 0.05) Different letters indicate significant differences between two methods 图 4 两种方法测定3种土壤的NO2-浓度 Figure 4 The concentrations of NO2- measured by both methods of colorimetry and chemiluminescence |
土壤待测液B与A的NO2-浓度之差为37.1~50.1 μg·L-1,其回收率RB-A为79.0%~90.7%(比色法)和93.1%~106.6%(NC法);土壤待测液B与C的NO2-浓度的差值为45.2~51.6 μg·L-1,比色法测定三种土壤NO2-浓度的回收率RC-B为92.7%~103.1%,NC法的回收率RC-B为90.3%~96.6%(表 2)。

|
依据Nagababu等[28]提供的测定血浆中NO2-浓度选用的反应剂(7 mL纯冰醋酸和1 mL 0.05 mol·L-1抗坏血酸),本研究选择较大的反应剂体积(增加2倍,以满足对高浓度NO2-样品的检测)和较低的冰醋酸浓度(50%,以缩短反应剂背景信号的稳定时间),即反应剂采用20 mL 50%的冰醋酸和3 mL 0.05 mol·L-1抗坏血酸混合。结果显示反应剂的背景NO信号值能够在2 h内稳定,而且连续重复测定10次50 μg·L-1 NaNO2标准溶液的NO信号峰面积稳定(CV为1.2%),表明50%冰醋酸提供的酸性条件不影响该浓度NO2-的还原效率,并可满足对500 ng N含量的NO2-测定。
3.2 NC法的精度本研究采用NC法,选择50%冰醋酸提供酸性条件,以0.05 mol·L-1抗坏血酸作为还原剂,测定NO2-标准溶液浓度,检测范围为2~500 μg·L-1,检出限为2 μg·L-1,即10 μg·kg-1(按水土比5:1计),在95%置信区间内的精度为1.2 μg·L-1。前人采用NC法主要用于测定水体和人体血浆中NO2-浓度(表 3),如Cox[22]测定天然水体的NO2-浓度,精度为0.4 μg·L-1;王燕等[24]测定水体中NO2-浓度的精度为0.34 μg·L-1;Nagababu等[25]测定血浆中NO2-浓度的精度为0.16 μg·L-1。本研究中NC法的检测精度为1.2 μg·L-1,高于前人采用NC法的测定结果。这可能是由于不同的研究者选择的氮氧化物分析仪、酸性条件与还原剂不同造成的。

|
不同的氮氧化物分析仪其测样频率和载气流速均不同。本研究采用热电环境仪器公司出品的氮氧化物分析仪(型号42i),其对NO浓度的检测频率最高为1 s一个数据,即1 Hz,远低于Nagababu等[28]采用一氧化氮分析仪(Sievers280i,通用电气分析仪器公司,美国)的采样频率4 Hz。两台仪器的检测限相近(1 nmol·mol-1左右),而高的采样频率会提高NO2-的检测精度。本研究所用的载气流速为500 mL·min-1,以满足热电环境仪器公司出品的氮氧化物分析仪的使用要求,但该载气流速高于之前研究采用的流速101~300 mL·min-1(表 3)。Bateman等[34]的研究表明,流速在42~126 mL·min-1范围时,加大流经反应瓶的载气流速,会促进反应瓶中的NO由液态向气态转化,从而加快NO气体与液体的分离速度,同时会减少NO在氮氧化物分析仪中与O3的反应时间,导致NO浓度信号值降低,但能提高方法精度;而王燕等[24]观测到随着载气流速的增加(150~750 mL·min-1),反应的信号值先增大后降低,当流速为480 mL·min-1时信号值最大。如果信号值高导致精度欠佳,则本研究采用大的载气流速可能是导致精度低的一个原因。但是对于本试验,降低载气流速是否能提高方法的精度,还有待于进一步验证。
此外,不同的研究者采用NC法测定NO2-浓度时选用不同物质作为还原剂和酸性环境,如Amini等[27]选择碘(I-)和硫酸,王燕等[24]选择矾(V3+)和盐酸。本研究之所以选择抗坏血酸作为还原剂,主要是因为其相对于碘和矾来说,能更好地避免硝酸盐、亚硝胺、亚硝基硫醇等物质的干扰[25],而这些物质在土壤中也普遍存在。至于不同还原剂和酸性条件是否会影响方法的检测精度仍需进一步研究。
3.3 NC法与比色法比较采用NC法测定三种不同酸碱度(pH 5.4~8.7)土壤中NO2-浓度的回收率结果较好,为90%~97%。本研究NC法测定土壤NO2-浓度的检出限和精度均优于比色法。对于酸性土壤(HN和HB)的NO2-,采用NC法与比色法的测定结果没有显著差异,但对于碱性土壤(SX)来说,NC法的测定结果较比色法显著提高11%~24%。这可能是由土壤性质的差异导致的。HN土壤的有机质和总氮含量明显高于HB土壤,但两方法测定HN和HB土壤的NO2-浓度结果无显著差异,表明有机质和总氮可能不是造成差异的原因。两方法测定结果差异显著的SX土壤,pH值为8.7,高于HN和HB土壤(5.4和6.2),属于碱性土壤。Amini等[27]采用NC法测定土壤浸提液中的NO2-浓度时发现,样品中Ca2+、SO42-和CO32-会对NO2-的测定产生一定的干扰,而碱性土壤中上述离子的含量一般较多。这可能是导致NC法测定SX土壤NO2-浓度较比色法偏高的一个原因。但由于本试验所用的土壤种类较少,土壤酸碱性对测定结果的影响有待后续增加样本数量来验证。
3.4 NC法的优缺点NC法对于土壤中NO2-浓度的测定有以下三方面优点:
(1)样品用量少。本试验中,考虑到土壤浸提液中NO2-浓度范围和操作方便等问题,采用进样量为1 mL;若待测液的NO2-浓度较大时,还可适当减少进样量。
(2)不受浸提液中悬浮物质或颜色的影响。而采用比色法测定NO2-浓度时,需对有悬浮物或有颜色的待测液进行前处理,过程复杂,费时费力。
(3)所需试剂容易配制,尤其针对比色法某些试剂含有毒性而言,操作简单安全。
但NC法较比色法也存在着不足:
(1)样品分析效率较低。本研究中NC法测定NO2-样品的时间为8~15 min·个-1,随着样品中NO2-浓度的增加,分析时间也相应增加,对于批量样品测定效率并不高。如对于40个NO2-样品( < 100 μg·L-1)的测定,采用比色法仅需1 h,而采用NC法至少需要6 h。
(2)系统稳定时间较长。比色法的显色仅需20 min,而NC法中待反应瓶背景NO信号值降低至5 nmol·mol-1以下并稳定至少需要2 h。
(3)对装置气密性要求很高,否则外界环境中NO的浓度会对测定造成影响,而比色法则不受环境中NO浓度的影响。
(4)仪器维护成本较高。NC法中使用的氮氧化物分析仪需定期进行标定(需要标定系统及标准气体等)及维护(更换颗粒过滤器及清洗仪器管路等),其成本明显高于比色法中分光光度计的维护成本。
4 结论本研究采用气相转化-化学发光法,选择50%冰醋酸提供酸性条件,以0.05 mol·L-1抗坏血酸作为还原剂,测定土壤中的NO2-含量。该方法检测范围为2~500 μg·L-1,平均变异系数为2.5%,在95%置信区间内测定精度为6 μg·kg-1(水土比为5:1),方法回收率为90%~97%。该方法测定酸性土壤NO2-含量与国标比色法无显著差异,但可能会高估碱性土壤的NO2-含量。气相化学发光法不受浸提液中悬浮物质或有色物质的影响,所用试剂无毒性,但分析时间随着NO2-浓度的增加而增长(8~15 min),限制了该方法的分析效率。后续研究可考虑通过增加不同类型土壤样本、更换不同的还原剂或降低反应流速,进一步评估该方法的适用性,并提高方法的精度和分析效率。
致谢 对本研究提供技术支持和帮助的有中国科学院大气物理研究所的付永锋、张寒、马磊和李来港等,谨致谢忱。| [1] |
Butterbach-Bahl K, Baggs E M, Dannenmann M, et al. Nitrous oxide emissions from soils:How well do we understand the processes and their controls?[J]. Philosophical Transactions of the Royal Society B, 2013, 368(1621): 20130122. DOI:10.1098/rstb.2013.0122 |
| [2] |
Fowler D, Pilegaard K, Sutton M A, et al. Atmospheric composition change:Ecosystems-atmosphere interactions[J]. Atmospheric Environment, 2009, 43(33): 5193-5267. DOI:10.1016/j.atmosenv.2009.07.068 |
| [3] |
Intergovermental Panel on Climate Change. Climate change 2013:The physical science basis, contribution of working groupⅠto the fifth assessment report[R]. Cambridge:Cambridge University Press, 2014.
|
| [4] |
Ravishankara A R, Daniel J S, Portmann R W. Nitrous Oxide(N2O):The dominant ozone-depleting substance emitted in the 21st Century[J]. Science, 2009, 326(5949): 123-125. DOI:10.1126/science.1176985 |
| [5] |
Su H, Cheng Y F, Oswald R, et al. Soil nitrite as a source of atmospheric HONO and OH radicals[J]. Science, 2011, 333(6049): 1616-1618. DOI:10.1126/science.1207687 |
| [6] |
Kulmala M, Petäjä T. Soil nitrites influence atmospheric chemistry[J]. Science, 2011, 333(6049): 1586-1587. DOI:10.1126/science.1211872 |
| [7] |
Cockburn A, Brambilla G, Fernández M L, et al. Nitrite in feed:From animal health to human health[J]. Toxicology and Applied Pharmacology, 2013, 270(3): 209-217. DOI:10.1016/j.taap.2010.11.008 |
| [8] |
Aschebrook-Kilfoy B, Shu X O, GaoY T, et al. Thyroid cancer risk and dietary nitrate and nitrite intake in the Shanghai Women's Health Study[J]. International Journal of Cancer, 2013, 132(4): 897-904. DOI:10.1002/ijc.27659 |
| [9] |
Della Valle C T, Daniel C R, Aschebrook-Kilfoy B, et al. Dietary intake of nitrate and nitrite and risk of renal cell carcinoma in the NIH-AARP Diet and Health Study[J]. British Journal of Cancer, 2013, 108: 205-212. DOI:10.1038/bjc.2012.522 |
| [9] |
Abdul Galil M S, Mahadevaiah, Yogendra Kumar M S, et al. A simple and rapid spectrophotometric method for the determination of nitrite by its decolorizing effect on peroxovanadate complex[J]. Spectrochimical Acta Part A, 2007, 67(1): 76-82. DOI:10.1016/j.saa.2006.06.026 |
| [11] |
Moldovan Z. Spectrophotometric determination of nitrite by its catalytic effect on the oxidation of congo red with bromate[J]. Bulletin of the Chemical Society of Ethiopia, 2012, 26(2): 159-169. |
| [12] |
Masserini Jr R T, Fanning K A. A sensor package for the simultaneous determination of nanomolar concentrations of nitrite, nitrate, and ammonia in seawater by fluorescence detection[J]. Marine Chemistry, 2000, 68(4): 323-333. DOI:10.1016/S0304-4203(99)00088-2 |
| [13] |
Menon S, Vikraman A E, Jesny S, et al. "Turn On" fluorescence determination of nitrite using green synthesized carbon nanoparticles[J]. Journal of Fluorescence, 2016, 26(1): 129-134. DOI:10.1007/s10895-015-1692-0 |
| [14] |
He D Y, Zhang Z J, Huang Y, et al. Chemiluminescence microflow injection analysis system on a chip for the determination of nitrite in food[J]. Food Chemistry, 2007, 101(2): 667-672. DOI:10.1016/j.foodchem.2006.02.024 |
| [15] |
Yaqoob M, Folgado Biot B, Nabi A, et al. Determination of nitrate and nitrite in freshwaters using flow-injection with luminol chemiluminescence detection[J]. Luminescence, 2012, 27(5): 419-425. DOI:10.1002/bio.v27.5 |
| [16] |
Kage S, Kudo K, Ikeda N. Simultaneous determination of nitrate and nitrite in human plasma by gas chromatography-mass spectrometry[J]. Journal of Analytical Toxicology, 2002, 26(6): 320-324. DOI:10.1093/jat/26.6.320 |
| [17] |
Stalikas C D, Konidari C N, Nanos C G. Ion chromatographic method for the simultaneous determination of nitrite and nitrate by post-column indirect fluorescence detection[J]. Journal of Chromatography A, 2003, 1002(1/2): 237-241. |
| [18] |
胡平, 解彦平, 任永红. 固相萃取-离子色谱法测定土壤中亚硝酸盐和硝酸盐[J]. 理化检验(化学分册), 2012, 48(11): 1318-1320. HU Ping, XIE Yan-ping, REN Yong-hong. IC determiniation of nitrite and nitrate in soil with solid phase extraction[J]. Physical Testing and Chemical Analysis Part B:Chemical Analysis, 2012, 48(11): 1318-1320. |
| [19] |
Suvardhan K, Suresh kumar K, Hari babu S, et al. RETRACTED:Simultaneous flow-through determination of nitrites, nitrates and their mixtures in environmental and biological samples using spectrophotometry[J]. Talanta, 2005, 66(2): 505-512. DOI:10.1016/j.talanta.2004.11.025 |
| [20] |
López Pasquali C E, Gallego-Picó A, Fernández Hernando P, et al. Two rapid and sensitive automated methods for the determination of nitrite and nitrate in soil samples[J]. Microchemical Journal, 2010, 94(1): 79-82. DOI:10.1016/j.microc.2009.09.005 |
| [21] |
国家环境保护部. GB 7493-1987水质, 亚硝酸盐氮的测定, 分光光度法[S]. 北京: 中国标准出版社, 1987. Ministry of Environmental Protection of the Perple's Republic of China. GB 7493-1987, Water quality, determination of nitrigen(nitrite), spectrophotometric method[S]. Beijing:China Standards Press, 1987. http://d.wanfangdata.com.cn/Standard_ISO%206777-1984.aspx |
| [22] |
Cox R D. Determination of nitrate and nitrite at the parts per billion level by chemiluminescence[J]. Analytical Chemistry, 1980, 52(2): 332-335. DOI:10.1021/ac50052a028 |
| [23] |
Braman R S, Hendrix S A. Nanogram nitrite and nitrate determination in environmental and biological materials by vanadium(Ⅲ) reduction with chemiluminescence detection[J]. Analytical Chemistry, 1989, 61(24): 2715-2718. DOI:10.1021/ac00199a007 |
| [24] |
王燕, 刘素美, 任景玲, 等. 化学发光法测定天然水体中低浓度硝酸盐和亚硝酸盐的含量[J]. 海洋科学, 2011, 35(5): 95-99. WANG Yan, LIU Su-mei, REN Jing-ling, et al. Chemiluminescent measurement of low concentrations of nitrate and nitrite[J]. Marine Sciences, 2011, 35(5): 95-99. |
| [25] |
Nagababu E, Rifkind J M. Measurement of plasma nitrite by chemiluminescence without interference of S-, N-nitroso and nitrated species[J]. Free Radical Biology and Medicine, 2007, 42(8): 1146-1154. DOI:10.1016/j.freeradbiomed.2006.12.029 |
| [26] |
张桂玲. 中国近海部分海域溶解甲烷和氧化亚氮的生物地球化学研究[D]. 青岛: 中国海洋大学, 2004: 24-28. ZHANG Gui-ling. Studies on biogeochemistry of dissolved methane and nitrous oxide in the coastal waters of China[D]. Qingdao:Ocean University of China, 2004:24-28. http://cdmd.cnki.com.cn/Article/CDMD-10423-2004131348.htm |
| [27] |
Amini M K, Pourhossein M, Talebi M A. Chemiluminescence flow injection system for nitrite ion determination[J]. Journal of the Iranian Chemical Society, 2005, 2(4): 305-314. DOI:10.1007/BF03245935 |
| [28] |
Nagababu E, Rifkind J M. Measurement of plasma nitrite by chemiluminescence[J]. Methods in Molecular Biology, 2010, 610: 41-49. DOI:10.1007/978-1-60327-029-8 |
| [29] |
Liu C Y, Wang K, Meng S X, et al. Effects of irrigation, fertilization and crop straw management on nitrous oxide and nitric oxide emissions from a wheat-maize rotation field in Northern China[J]. Agriculture, Ecosystems and Environment, 2011, 140(1/2): 226-233. |
| [30] |
Yao Z, Du Y, Tao Y, et al. Water-saving ground cover rice production system reduces net greenhouse gas fluxes in an annual rice-based cropping system[J]. Biogeosciences, 2014, 11: 6221-6236. |
| [31] |
Shen J L, Tang H, Liu J Y, et al. Contrasting effects of straw and straw-derived biochar amendments on greenhouse gas emissions within double rice cropping systems[J]. Agriculture, Ecosystems and Environment, 2014, 188: 264-274. DOI:10.1016/j.agee.2014.03.002 |
| [32] |
Burakham R, Oshima M, Grudpan K, et al. Simple flow-injection system for the simultaneous determination of nitrite and nitrate in water samples[J]. Talanta, 2004, 64(5): 1259-1265. DOI:10.1016/j.talanta.2004.03.059 |
| [33] |
曹娜, 王睿, 廖婷婷, 等. 厌氧条件下砂壤水稻土N2、N2O、NO、CO2和NH4排放特征[J]. 环境科学, 2015, 36(9): 3373-3382. CAO Na, WANG Rui, LIAO Ting-ting, et al. Characteristics of N2, N2O, NO, CO2 and NH4 emissions in anaerobic condition from sandy loam paddy soil[J]. Environmental Science, 2015, 36(9): 3373-3382. |
| [34] |
Bateman R M, Ellis C G, Freeman D J. Optimization of nitric oxide chemiluminescence operating conditions for measurement of plasma nitrite and nitrate[J]. Clinical Chemistry, 2002, 48(3): 570-573. |