 2019, Vol. 38
2019, Vol. 38


2. 西南石油大学工业危废处置与资源化利用研究院, 成都 610500
2. Institute of Industrial Hazardous Waste Disposal and Utilization, Southwest Petroleum University, Chengdu 610500, China
石油在开采、加工、运输及储藏等过程中存在的非正常泄漏,导致石油类物质进入土壤,破坏土壤结构,改变其物理化学性质,影响农作物的品质,甚至危害人类健康[1-2]。石油污染土壤需及时进行修复处理,原位化学氧化(ISCO)是重要的修复手段之一[3-6]。ISCO通过向污染土壤注入强氧化剂进而氧化降解有机污染物,使得污染物减量或是毒性降低更易被微生物利用而降解[7-8]。
ISCO技术最常用的几种氧化剂是H2O2、KMnO4、Na2S2O8[9-13]。其中H2O2和Na2S2O8需要利用土壤本身的铁、锰矿物作为催化剂,或是外加催化剂,活化生成·OH、SO4-·,才能有效氧化降解大部分有机污染物;KMnO4可直接氧化不饱和脂肪族化合物,但对芳香族化合物、氯烷烃类污染物氧化效率很低,产生的二氧化锰沉淀会堵塞土壤孔隙影响氧化剂的传输[14-15]。由于土壤的异质性和低扩散性,使得氧化剂在土壤中的扩散传递受到限制。在扩散过程中,H2O2在土壤矿物和微生物作用下极易分解生成O2而快速失去化学氧化作用[16],半衰期仅为数分钟到数小时,而Na2S2O8和KMnO4则相对稳定,半衰期可达数月[17]。Chen等[18]研究了不同氧化剂(H2O2、Na2S2O8、KMnO4)在剂量为1%~10%(W/W)时修复柴油污染土壤,指出柴油去除率与氧化剂在土壤中的存留时间(Na2S2O8 > KMnO4 > H2O2)成正比。低浓度分批投加氧化剂H2O2,变相延长氧化剂的存留时间,可以显著增加对土壤中有机污染物的去除率[19-20]。因此,氧化剂本身或是其生成的活性氧化物质(ROS)的氧化能力,以及氧化剂保持有效浓度的时间和扩散范围,是影响ISCO对土壤有机污染修复效率的重要因素。
本研究针对ISCO中氧化剂的持续有效性,通过室内实验,以H2O2、Na2S2O8为氧化剂,将总量相等的氧化剂1次性或分3次加入到石油污染土壤中,在有或无外源催化剂(零价铁ZVI)添加下,监测反应体系中氧化剂剩余量及总石油烃类(TPH)含量随时间的变化,研究比较不同氧化剂及添加方式对氧化剂持续有效性、TPH去除速率和最终去除率的影响,探讨基本反应机理,为化学氧化修复土壤石油污染提供一定的理论支持。
1 材料与方法 1.1 试剂和材料过硫酸钠(Na2S2O8)、过氧化氢(H2O2)、铁粉(ZVI)、硅镁吸附剂、碘化钾、无水硫酸钠、丙酮、正己烷、二氯甲烷均为分析纯,购自成都市科龙化工试剂厂。四氯化碳,优级纯,购自天津傲然精细化工研究所。实验用水均为超纯水。
土壤样品采自西南石油大学三期东面林地土,拣出植物残体、石子等杂物,经风干、磨细、过筛(0.5 mm孔径),避光保存待用。
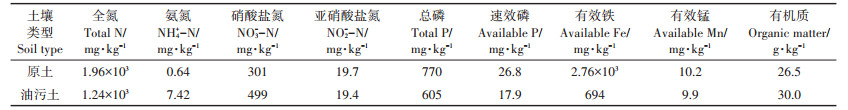
1.2 石油污染土壤的制备定量称取5 g石油原油(取自辽河油田某井),加入100 mL四氯化碳,搅拌混合均匀后,加入到1 kg土样中,边加边搅拌,再以100 mL四氯化碳分次冲洗烧杯,加入土壤中,搅拌均匀后置于通风厨中24 h,至四氯化碳挥发殆尽,制得石油污染土壤。测得其总石油烃含量为(4 632.7±200)mg·kg-1。供试土壤样品基本理化性质如表 1。
|
|
表 1 供试土壤基本理化性质 Table 1 Physico-chemical properties of the tested soil |
以150 mL锥形瓶为反应器,依次加入上述石油污染土壤(5 g)、去离子水(5 mL)、氧化剂(5 mL),控制体系固液比为1:2(g:mL),摇匀后以保鲜膜封口,以减少水分损失。每组均设置3个平行样,室温静置反应,分别在第0、2、4、6、10 d取样,测定溶液pH值、剩余氧化剂浓度,以及土壤中TPH含量。
不同实验处理:(1)一次投加氧化剂Na2S2O8或H2O2,溶液体系中初始浓度为21、63、105 mmol·L-1;(2)以铁粉为活化剂,Na2S2O8在溶液体系中初始浓度为21、63、105 mmol·L-1;(3)H2O2分3次投加,控制H2O2初始浓度总量分别为21、63、105 mmol·L-1,在反应第0、24、48 h分3次等量投加。对照(CK)处理以添加等体积的去离子水代替氧化剂,氧化剂均现配并以相应浓度添加,150 mg ZVI预先加入250 g污染土样并尽可能混匀备用。
1.4 样品处理和分析测定 1.4.1 氧化剂提取与测定将锥形瓶内石油污染土壤泥浆转移至50 mL离心管,分两次共加入10 mL纯水清洗锥形瓶,清洗液转移至离心管,保证土壤样品尽可能完全转移。6000 r·min-1离心10 min,上层液体倒入另一离心管。再加入10 mL纯水至含泥浆离心管,振荡,摇匀,再次离心,收集上层液体,重复以上实验步骤两次,得到上清液约40 mL,真空抽滤至50 mL比色管,纯水定容,用于测定剩余氧化剂浓度。氧化剂含量采用碘量法测定[21]。
1.4.2 TPH提取与测定离心管中泥浆样品转移至坩埚中,以10 mL纯水分两次清洗离心管,清洗液转移至坩埚,50 ℃低温烘干,磨细后用于测定TPH含量。以四氯化碳为浸提剂,定量称取1 g石油污染土壤干样,置于离心管内,加入10 mL四氯化碳,超声清洗15 min,6000 r·min-1离心10 min,提取液转入50 mL离心管,采用四氯化碳重复提取3次。提取液真空抽滤至50 mL比色管,四氯化碳定容,加入5 g硅镁吸附剂以除去植物油,再次抽滤后收集滤液,采用红外测油仪测定其中TPH浓度,再换算为土壤中剩余TPH含量。
1.4.3 土壤样品有机成分GC-MS分析称取3 g土壤干样于离心管中,加入30 mL正己烷、丙酮混合液(体积比1:1),盖紧摇匀,超声提取30 min,再置于摇床振荡12 h。6000 r·min-1离心10 min,过滤(加入5 g无水硫酸钠)至锥形瓶中;氮吹仪浓缩至近干,并将溶剂置换为二氯甲烷;用2 mL二氯甲烷进行浓缩,并通过0.22 μm有机针孔滤膜至浓缩瓶中,待分析[22]。
GC-MS分析条件:毛细管色谱柱,HP-5MS(30 m×0.25 mm×0.25 μm);进样方式,自动;流量,1 mL· min-1;载气,He;分流比,40:1;初始温度,280 ℃;进样口温度,280 ℃;进样量,0.8 μL;溶剂延迟,4 min。程序升温:初始温度为40 ℃,然后以5 ℃·min-1升到280 ℃,保留5 min。扫描范围:全扫描。
1.5 数据分析采用Excel 2007和Origin 8.0单因素方差分析(ANOVA)对试验数据进行统计、分析。
2 结果与分析 2.1 不同氧化剂在土壤中的持续有效性分析 2.1.1 Na2S2O8对TPH去除的持续有效性如图 1所示,在3种初始浓度21、63、105 mmol· L-1处理下,Na2S2O8剩余量在反应前4 d快速降低;初始Na2S2O8浓度越高,Na2S2O8分解量越多,但分解率相对越低,依次为54.76%、50.40%、47.38%。随时间增加,反应体系中Na2S2O8剩余量降低速率减慢,最终趋于平稳。在未加入氧化剂的CK处理中,TPH含量也有一定程度的降低(10 d降解率为8.44%),可能与土壤中的微生物作用有关。加入Na2S2O8的处理中,TPH含量在反应前4 d均快速减少,但在第4~10 d TPH去除量增加较微弱。反应期间Na2S2O8分解速率与TPH降解速率变化趋势的高度一致性,表明TPH的降解与Na2S2O8的分解具有明显相关关系。此外,反应10 d后Na2S2O8剩余量仍较充足,体系中Na2S2O8剩余量分别为11、35、60 mmol·L-1,表明去除TPH的限制因子应该是土壤中具有催化活性的物质不足。即Na2S2O8在土壤中的存留具有一定的持续性,但催化活性物质不足限制了Na2S2O8对TPH去除的有效性。因此,下文以铁粉为外加催化剂进行研究。

|
图 1 一次投加Na2S2O8处理中Na2S2O8剩余量(a)和TPH含量变化(b) Figure 1 Residual amount of Na2S2O8(a)and TPH content (b)changes while adding Na2S2O8 at one time |
3种初始浓度处理中,H2O2在反应第2 d均已检测不到(图 2),表明其快速分解,在土壤中存留时间短,持续性较差。但在反应第2~10 d,与CK组相比,TPH含量仍有明显的降低,H2O2初始浓度为21、63、105 mmol·L-1时,TPH降解率增加量分别为26.24%、18.63%、29.93%。尽管H2O2在反应前2 d已耗尽,但TPH在第2~10 d仍持续有一定的降解,这可能与后续微生物作用去除TPH有关。H2O2处理使得石油烃类物质中难降解成分被氧化后更易于微生物降解[23],且反应体系中H2O2快速分解消耗,对土壤中微生物的持续抑制作用较弱。在中、高初始浓度(63、105 mmol·L-1)H2O2的处理中,TPH在2~10 d的去除率增加量略低于21 mmol·L-1的处理,可能与高浓度氧化剂快速释放ROS对微生物有较强的灭活作用有关,对后续微生物修复的不利影响更大[18]。H2O2分解过快和高初始浓度对微生物的抑制作用使得TPH总去除率偏低,下一步通过少量多次投加H2O2对TPH去除进行研究。

|
图 2 一次投加H2O2处理中H2O2剩余量(a)和TPH含量变化(b) Figure 2 Residual amount of H2O2(a)and TPH content (b)changes while adding H2O2 at one time |
添加ZVI活化Na2S2O8反应体系中,Na2S2O8剩余量和TPH含量变化如图 3所示。在反应前4 d,Na2S2O8剩余量快速降低,初始Na2S2O8浓度越高,其分解量越多;在Na2S2O8初始浓度为21、63、105 mmol·L-1时的分解率分别为75.6%、61.1%、55.5%。与不加ZVI的处理(图 1)相比,加入ZVI后Na2S2O8分解率在不同初始浓度下均明显增加。反应第4~10 d,体系中Na2S2O8浓度变化均较小。反应10 d后Na2S2O8剩余量分别为4.83、24.50、46.88 mmol·L-1,比未加ZVI处理的消耗量分别增加了4.67、6.75、8.37 mmol·L-1。

|
图 3 ZVI活化Na2S2O8处理中Na2S2O8剩余量(a)和TPH含量(b)变化 Figure 3 Residual amount of Na2S2O8(a)and TPH content (b)changes while adding Na2S2O8 with ZVI activation |
加入ZVI催化剂活化Na2S2O8的处理组中,Na2S2O8的浓度越高,TPH降解率越高。Na2S2O8初始浓度为21、63、105 mmol·L-1时,在反应前6 d TPH含量均快速下降,降解率分别为50.78%、58.69%、51.98%。第6~10 d,TPH去除率增加不明显,与Na2S2O8浓度变化趋势一致。结果表明,在添加外源催化剂ZVI后,反应体系中Na2S2O8的分解速率和终分解量都得到一定程度的提升,且TPH降解率也明显增加,但对于高浓度Na2S2O8处理石油污染土壤时,ZVI用量仍不足以完全活化Na2S2O8。
2.3 分批投加H2O2对土壤中TPH的降解如下图 4所示,H2O2初始浓度总量为21、63、105 mmol·L-1,分3次在反应第0、24、48 h投加,土壤中TPH含量在第0~4 d内快速降低,4 d后降解率分别为54.40%、57.08%、59.48%,略高于一次投加的处理(图 2)。分次投加可增加H2O2在反应体系中的存留时间,使得H2O2与石油烃类污染物充分接触,一定程度提高了TPH的氧化去除率。

|
图 4 H2O2分3次投加处理中H2O2剩余量(a)和TPH含量(b)变化 Figure 4 Residual amount of H2O2(a)and TPH content (b)changes while adding H2O2 in three portions |
石油烃类组分复杂,GC-MS法能够比传统方法获得更多的烃类组成信息。以105 mmol·L-1氧化剂处理前及反应10 d的石油污染土壤为代表,石油烃成分GC-MS图谱分析如图 5所示(图中标注主要峰)。氧化处理10 d后,石油烃类各成分含量均有明显变化,GC-MS图谱总峰值在H2O2分批次处理、ZVI活化Na2S2O8处理中分别降低了56.64%和57.60%。其中处理前长链石油烃类主要成分为7-甲基十八烷、正十九烷、正三十烷,含量分别为7.57%、7.24%、6.90%。ZVI活化Na2S2O8处理后主要成分变为正十六烷、正十七烷、正十八烷,含量分别为11.61%、9.00%、6.88%;H2O2分批次处理后主要成分变为正十五烷、正十六烷、正十七烷,含量分别为14.12%、12.22%、8.90%。结果表明,主要成分中长链烷烃有变较短链烷烃的趋势,且使氧化处理后石油烃类主要组分中正十五烷、正十六烷、正十七烷等的比例明显增加。

|
图 5 化学氧化处理前后GC-MS图谱 Figure 5 GC-MS spectra before and after chemical oxidation treatment |
反应10 d后,在氧化剂初始浓度为63、105 mmol· L-1时,TPH总去除率在Na2S2O8处理中(46.20%、47.95%)显著高于H2O2处理(33.79%、33.75%),但在氧化剂初始浓度为21 mmol·L-1时,TPH总去除率在Na2S2O8和H2O2处理中无显著差异(P > 0.05),分别为38.09%、37.63%。H2O2和Na2S2O8处理下TPH在第0~ 4 d和4~10 d降解率变化如图 6所示,并对降解率进行差异显著性分析(P < 0.05)。在第0~4 d,TPH去除率在Na2S2O8处理中显著高于H2O2处理,氧化剂浓度为21、63、105 mmol·L-1时分别高出5.25%、21.01%、21.34%;在4~10 d,氧化剂初始浓度为21、63、105 mmol · L-1时,Na2S2O8处理中TPH去除率增加量(1.39%、2.16%、1.25%)显著低于H2O2处理(6.18%、10.76%、8.42%)。本研究反应体系对TPH的降解作用主要来自氧化剂的化学氧化,以及可能存在部分的微生物作用,后者则受到TPH的可生物利用性和氧化剂对微生物的抑制作用的影响[24]。

|
不同小写字母表示同一浓度、同一时间段的不同处理间差异显著(P < 0.05)。下同 Different lowercase letters indicate significant differences between treatments at the same concentration and in the same time period(P < 0.05). The same below 图 6 不同氧化剂处理下总石油烃降解率的变化 Figure 6 Changes of TPH degradation rate under different oxidant treatments |
在第0~4 d TPH快速降解,主要为氧化剂氧化作用贡献,因为此过程中TPH降解与氧化剂的浓度快速降低几乎同步。但与Na2S2O8处理相比,H2O2在土壤中存留时间更短,且易被土壤中矿物和微生物作用直接分解为O2,失去对有机物的氧化作用[16],不利于TPH的有效降解。
在上文指出H2O2处理中TPH在4~10 d内的持续降解可能与微生物作用有关,因为H2O2在2 d内快速消耗殆尽(图 2a),对微生物不具备持续抑制作用。同时,H2O2处理可将TPH中难降解的复杂大分子物质转化为相对易被生物降解的小分子化合物,从而促进后续微生物降解TPH,体现了化学氧化与微生物联合效应。相反,在Na2S2O8初始浓度为63、105 mmol· L-1时,反应10 d内Na2S2O8均保持较高浓度(图 1a),对微生物可能产生持续抑制作用[25],且由于催化活性物质不足,化学氧化去除TPH缺乏持续有效性,综合使得Na2S2O8处理中TPH去除率在第4~10 d增加微弱。在Na2S2O8初始浓度为21 mmol·L-1处理中,反应10 d内Na2S2O8均保持较低浓度,可能对微生物的抑制作用较弱,因此TPH去除率在第4~10 d增加明显。同时,在实验过程中发现,Na2S2O8处理中反应体系pH值随反应时间推移快速由7.55降低到3.75左右,变化过大且偏较强酸性,因此不利于微生物作用去除TPH[26];而H2O2处理中的pH值在5.23~7.68,变化较小且偏中性条件,利于大多数微生物的繁殖[27]。
3.2 添加催化剂ZVI处理下TPH变化氧化剂初始浓度为21、63、105 mmol·L-1时,添加ZVI处理中TPH总去除率(53.39%、59.63%、64.71%)都显著高于未加入ZVI处理组(38.09%、46.20%、47.95%)(P < 0.05)。外加ZVI对Na2S2O8处理下TPH在第0~4 d和4~10 d降解率变化如图 7所示。在反应第0~4 d,TPH降解率在有无催化剂ZVI下无明显差异,但加入ZVI促进了反应第4~10 d TPH的持续降解,在Na2S2O8初始浓度为21、63、105 mmol·L-1处理中TPH降解率分别增加了8.46%、8.49%、12.26%。

|
图 7 有无ZVI活化Na2S2O8处理下TPH降解率的变化 Figure 7 Change of TPH degradation rate with or without ZVI activated Na2S2O8 treatments |
土壤中天然存在的过渡金属及其氧化物,能够一定程度活化Na2S2O8,因此在未加入催化剂的处理中,TPH含量有明显降解。但是土壤中铁、锰等对Na2S2O8具有催化活性的成分含量不足,且在与Na2S2O8反应后变为高价氧化态,在土壤中难以回到还原态,即失去催化活性。加入ZVI可以缓慢释放出还原态铁(零价铁和二价铁)[28],提高反应体系对Na2S2O8持续活化作用产生SO4·-(反应1和反应2),从而提高Na2S2O8氧化去除TPH的效果。酸性条件下,零价铁可与三价铁反应生成Fe2+(反应3)[29],使溶液中保持一定的Fe2+浓度,更利于活化Na2S2O8生成SO4-·。因此,加入ZVI提高了反应体系中催化剂的有效浓度和持续时间,大幅促进了Na2S2O8对TPH的去除率和持续去除效果。
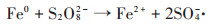
|
(1) |
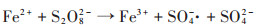
|
(2) |
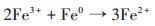
|
(3) |
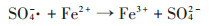
|
(4) |
在反应10 d后,氧化剂初始浓度为21、63、105 mmol·L-1时,TPH总去除率在分3次加入H2O2处理中(54.90%、59.22%、61.85%)显著高于一次加入H2O2处理(37.63%、33.79%、33.75%)(P < 0.05)。H2O2一次投加与分3次投加处理下TPH在第0~4 d和第4~10 d降解率变化如图 8所示。在反应第0~4 d,分批投加H2O2处理中TPH去除率显著增加(P < 0.05),在氧化剂初始浓度为21、63、105 mmol · L-1时,分别高出22.95%、34.05%、34.15%。但在反应第4~10 d,分3次投加处理中TPH降解率增加量均较小(分别为0.49%、2.14%、2.38%),远低于一次投加的处理(分别为9.02%、13.97%、11.27%)。

|
图 8 H2O2在不同投加方式下TPH降解率的变化 Figure 8 Change of TPH degradation rate of H2O2 under different dosage modes |
分次投加增加了H2O2在反应体系中的存留时间和利用率[30-31],进而提高了对TPH氧化去除率。但分批在第0、24、48 h内加入H2O2,造成的持续干扰可能会更大程度抑制或灭活土壤中的微生物,使得在第4~10内TPH含量变化不明显。而一次投加H2O2的处理,H2O2在反应体系中分解非常快,2 d内(甚至更短)即消耗殆尽,在第4~10 d可能存在较强的微生物作用,能够持续降解TPH。
4 结论(1)Na2S2O8在石油污染土壤中存留时间长,具有较好的持续性。但土壤本身含有的催化活性物质不足,使得Na2S2O8对TPH去除的有效性不足。加入ZVI显著增加了TPH在第4~10 d的持续降解和总降解率,氧化剂初始浓度为21、63、105 mmmol·L-1时,TPH降解率较未加入ZVI处理组分别高出8.46%、8.49%、12.26%。
(2)H2O2分次加入可增加其在反应体系中的有效存留时间,提高对TPH去除的持续性。在反应10 d后,氧化剂初始浓度为21、63、105 mmol·L-1时,分批次加入H2O2对TPH的降解率分别提高了17.27%、25.43%、28.11%。
(3)低浓度Na2S2O8和一次投加H2O2的处理中,可能存在一定的微生物作用,使得TPH在反应后期具有一定的持续降解;相反,在高浓度Na2S2O8和分3次投加H2O2的处理中,则可能由于氧化剂对微生物的持续抑制作用,未能体现微生物作用对TPH的持续降解。
| [1] |
杜卫东, 万云洋, 钟宁宁, 等. 土壤和沉积物石油污染现状[J]. 武汉大学学报(理学版), 2011, 57(4): 311-322. DU Wei-dong, WAN Yun-yang, ZHONG Ning-ning, et al. Current status of petroleum-contaminated soil and sediments[J]. Journal of Wuhan University, 2011, 57(4): 311-322. |
| [2] |
王威, 苏小四, 张玉玲, 等. 石油类污染场地的自然衰减作用[J]. 吉林大学学报(地球科学版), 2011, 41(增刊1): 310-314, 327. WANG Wei, SU Xiao-si, ZHANG Yu-ling, et al. Natural attenuation of a petroleum contaminated site[J]. Journal of Jilin University(Earth Science Edition), 2011, 41(Suppl 1): 310-314, 327. |
| [3] |
冯俊生, 张俏晨. 土壤原位修复技术研究与应用进展[J]. 生态环境学报, 2014, 23(11): 1861-1867. FENG Jun-sheng, ZHANG Qiao-chen. A review on the study on practice of soil remediation in situ[J]. Journal of Eco-Environment, 2014, 23(11): 1861-1867. DOI:10.3969/j.issn.1674-5906.2014.11.021 |
| [4] |
吴昊, 孙丽娜, 王辉, 等. 活化过硫酸钠原位修复石油类污染土壤研究进展[J]. 环境化学, 2015, 34(11): 2085-2095. WU Hao, SUN Li-na, WANG Hui, et al. Persulfate In-situ remediation of petroleum hydrocarbon contaminated soil[J]. Environmental Chemistry, 2015, 34(11): 2085-2095. DOI:10.7524/j.issn.0254-6108.2015.11.2015052601 |
| [5] |
燕启社, 孙红文. Fenton氧化对土壤有机质及其吸附性能的影响[J]. 农业环境科学学报, 2006, 25(2): 141-146. YAN Qi-she, SUN Hong-wen. Influence of Fenton oxidation on soil organic matter and its sorption characteristics[J]. Journal of Agro-Environment Science, 2006, 25(2): 141-146. |
| [6] |
赵丹, 廖晓勇, 阎秀兰, 等. 不同化学氧化剂对焦化污染场地多环芳烃的修复效果[J]. 环境科学, 2011, 32(3): 849-856. ZHAO Dan, LIAO Xiao-yong, YAN Xiu-lan, et al. Chemical oxidation for remediation of soils contaminated with polycyclic aromatic hydrocarbons at a coking site[J]. Environmental Science, 2011, 32(3): 849-856. |
| [7] |
Tsitonaki A, Petri B, Crimi M, et al. In situ chemical oxidation of contaminated soil and groundwater using persulfate:A review[J]. Critical Reviews in Environmental Science and Technology, 2010, 40(1): 55-91. DOI:10.1080/10643380802039303 |
| [8] |
Innocenti I, Verginelli I, Massetti F, et al. Pilot-scale ISCO treatment of a MTBE contaminated site using a Fenton-like process[J]. Science of the Total Environment, 2014, 485/486: 726-738. DOI:10.1016/j.scitotenv.2014.01.062 |
| [9] |
Ranc B, Faure P, Croze V, et al. Selection of oxidant doses for in situ chemical oxidation of soils contaminated by polycyclic aromatic hydrocarbons(PAHs):A review[J]. Journal of Hazardous Materials, 2016, 312: 280-297. DOI:10.1016/j.jhazmat.2016.03.068 |
| [10] |
Watts R J, Teel A L. Treatment of contaminated soils and groundwater using in situ chemical oxidation[J]. Hazardous Waste Management, 2006, 10(1): 2-9. |
| [11] |
Watts R J, Teel A L. Chemistry of modified Fenton's reagent(catalyzed H2O2 propagations-CHP) for in situ soil and ground water remediation[J]. Journal of Environmental Engineering, 2005, 131(4): 612-622. DOI:10.1061/(ASCE)0733-9372(2005)131:4(612) |
| [12] |
Corbin J F, Teel A L, Allen-King M, et al. Reactive oxygen species responsible for the enhanced desorption of dodecane in modified Fenton's systems[J]. Water Environment Research, 2007, 79(1): 37-42. DOI:10.2175/106143006X136793 |
| [13] |
Trantnyek P G, Waldemer R H. Kinetics of contaminant degradation by permanganate[J]. Environmental Science & Technology, 2006, 40(3): 1055-1061. |
| [14] |
Kakosová E, Hrabák P, Černík M, et al. Effect of various chemical oxidation agents on soil microbial communities[J]. Chemical Engineering Journal, 2017, 314: 257-265. DOI:10.1016/j.cej.2016.12.065 |
| [15] |
Yen C H, Chen K F, Kao C M, et al. Application of persulfate to remediate petroleum hydrocarbon-contaminated soil:Feasibility and comparison with common oxidants[J]. Journal of Hazardous Materials, 2011, 186(2/3): 2097-2102. |
| [16] |
Gates D D, Siegrist R L, Cline S R. Chemical oxidation of volatile and semi-volatile organic compounds in soil[C]//Proceedings of the 88th Annual Air and Waste Management Association Conference, San Antonio, Texas, 1995.
|
| [17] |
Liang C J, Chien Y C, Lin Y L. Impacts of ISCO persulfate, peroxide and permanganate oxidants on soils:Soil oxidant demand and soil properties[J]. Soil and Sediment Contamination:An International Journal, 2012, 21(6): 701-719. DOI:10.1080/15320383.2012.691129 |
| [18] |
Chen K F, Chang Y C, Chiou W T. Remediation of diesel-contaminated soil using in situ chemical oxidation(ISCO)and the effects of common oxidants on the indigenous microbial community:A comparison study[J]. Journal of Chemical Technology & Biotechnology, 2016, 91(6): 1877-1888. |
| [19] |
陈丽娜. Fenton和K2S2O8法处理五氯酚污染土壤实验研究[D].哈尔滨: 哈尔滨工程大学, 2009. CHEN Li-na. Study on degradation of PCP contaminated soil using Fenton and K2S2O8 oxidation[D]. Harbin: Harbin Engineering University, 2009. |
| [20] |
Xu J L, Deng X, Cui Y W, et al. Impact of chemical oxidation on indigenous bacteria and mobilization of nutrients and subsequent bioremediation of crude oil-contaminated soil[J]. Journal of Hazardous Materials, 2016, 320: 160-168. DOI:10.1016/j.jhazmat.2016.08.028 |
| [21] |
姚成, 许艳, 胡燕斌, 等. 碘量法测定双氧水的含量[J]. 江苏化工, 1996(3): 51-53. YAO Cheng, XU Yan, HU Yan-bin, et al. Determination of hydrogen peroxide by iodometric method[J]. Jiangsu Chemical Industry, 1996(3): 51-53. |
| [22] |
魏子明, 张文斌. 超声提取-GC/MS法测定土壤中的半挥发性有机物[J]. 建筑工程技术与设计, 2017(9): 745, 1685. WEI Zi-ming, ZHANG Wen-bin. Ultrasonic extraction, GC/MS method for determination of semi volatile organic compounds in the soil[J]. Construction Engineering Technology and Design, 2017(9): 745, 1685. |
| [23] |
魏德洲, 秦煜民. H2O2在石油污染土壤微生物治理过程中的作用[J]. 中国环境科学, 1997, 17(5): 429-432. WEI De-zhou, QIN Yu-min. Function of H2O2 in the bioremediation process of petroleum contaminated soil[J]. Chinese Environmental Science, 1997, 17(5): 429-432. DOI:10.3321/j.issn:1000-6923.1997.05.011 |
| [24] |
马强, 林爱军, 马薇, 等. 土壤中总石油烃污染(TPH)的微生物降解与修复研究进展[J]. 生态毒理学报, 2008, 3(1): 1-8. MA Qiang, LIN Ai-jun, MA Wei, et al. A review of micro-biological degradation and remediation of total petroleum hydrocarbon in soil[J]. Journal of Ecotoxicology, 2008, 3(1): 1-8. |
| [25] |
Yang S Y, Wang P, Yang X, et al. A novel advanced oxidation process to degrade organic pollutants in wastewater:Microwave-activated persulfate oxidation[J]. Journal of Environmental Sciences, 2009, 21(9): 1175-1180. DOI:10.1016/S1001-0742(08)62399-2 |
| [26] |
Kemmitt S J, Wright D, Goulding K W, et al. pH regulation of carbon and nitrogen dynamics in two agricultural soils[J]. Soil Biology and Biochemistry, 2006, 5(5): 898-911. |
| [27] |
Imtyaz Hussain.基于硫酸根自由基的高级氧化技术对典型有机污染物降解的研究[D].广州: 华南理工大学, 2013. Imtyaz Hussain. The study of sulfate-radicals based on advanced oxidation processes for the degradation of typical organic compounds[D]. Guangzhou: South China University of Technology, 2013. http://cdmd.cnki.com.cn/Article/CDMD-10561-1013320310.htm |
| [28] |
Liang C J, Lai M C. Trichloroethylene degradation by zero valent iron activated persulfate oxidation[J]. Environmental Engineering Science, 2008, 25(7): 1071-1077. DOI:10.1089/ees.2007.0174 |
| [29] |
Cullen L G, Tilston E L, Mitchell G R, et al. Assessing the impact of nano- and micro-scale zero valent iron particles on soil microbial activities:Particle reactivity interferes with assay conditions and interpretation of genuine microbial effects[J]. Chemosphere, 2011, 82(11): 1675-1682. DOI:10.1016/j.chemosphere.2010.11.009 |
| [30] |
Chu W, Chan K H, Kwan C Y, et al. Degradation of atrazine by modified stepwise-Fenton' s processes[J]. Chemosphere, 2007, 67(4): 755-761. DOI:10.1016/j.chemosphere.2006.10.039 |
| [31] |
Zhang H, Choi H J, Huang C. Optimization of Fenton process for the treatment of landfill leachate[J]. Journal of Hazardous Materials, 2005, 125(1): 166-174. |