 2019, Vol. 38
2019, Vol. 38



2. 中国环境科学研究院, 环境基准与风险评估国家重点实验室, 北京 100012
2. State Key Laboratory of Environmental Criteria and Risk Assessment, Chinese Research Academy of Environmental Sciences, Beijing 100012, China
在过去的十多年间,纳米材料在更小的尺度、更多样化的形貌以及更丰富的功能上都取得了巨大的进步,满足了人类对新材料、新功能的要求。二氧化钛纳米颗粒(TiO2 NPs)作为使用最广泛的纳米材料之一,由于其稳定的物理化学性能,具有均一的孔径、较小的尺寸、较高的比表面积、优良的光学性能以及催化性能,使得它广泛运用于如牙膏、防晒霜、光催化剂、化妆品、油漆涂料、太阳能电池设备等[1-3]。另外,TiO2 NPs由于其拥有较大的比表面积及更好的分散性,被应用于有机污染物的光催化降解,如对金属污染物如二价铅和二价镉的去除等水体环境的修复治理[4-5]。伴随着纳米材料的大量生产和使用其不可避免地进入自然环境,参与地球化学循环过程,并通过食物链在生物体中累积,对生态环境和人体健康产生潜在的危害[6]。已有研究报道多瑙河水体中TiO2 NPs浓度为0.6~4.7 mg·L-1[7],而TiO2 NPs进入到水体环境后,pH值、离子强度等因素能够改变TiO2 NPs表面电荷性质和强度而影响TiO2 NPs在水环境中的悬浮/沉降[6, 8]。而天然有机质(NOM)不仅能够直接吸附到TiO2 NPs表面,还能够与水环境中的其他污染物结合,进而对纳米材料和污染物产生双重影响。已有报道指出TiO2 NPs在迁移转化过程中,其毒理学性质会随着与其他物质的相互作用以及水体理化参数而改变[9-12]。因此,TiO2 NPs进入水体环境后受到水体理化要素的影响以及与其污染物的相互作用也成为研究者关注的焦点。
腐植酸(Humic acid,HA)是天然有机质重要的组成部分,腐植酸是大分子的、带负电荷的聚电解质,其组成和结构十分复杂,极大地影响着TiO2 NPs在水体环境中的环境行为[13]。HA富含羧基和酚基等多种官能团,可通过这些官能团与水体环境中的纳米材料相互作用。有研究表明HA对富勒烯(C60)的聚集动力学有着显著的影响,而HA的羧基或者酚基官能团能够与TiO2、SiO2、Al2O3和ZnO等纳米无机氧化物发生静电吸引、离子交换、疏水、络合等多种作用[14-19]。HA吸附到纳米颗粒表面,会使得它们结合后毒理学性质发生改变。有研究表明腐植酸与TiO2 NPs相互作用后对斑马鱼发育具有更大的影响[20]。同时,TiO2 NPs表面结合HA会抑制藻类细胞的黏附,从而减轻藻类细胞的氧化应激[21]。因此,在水环境中HA对纳米颗粒的环境行为和生态效应的影响不容忽视。
HA的组成和结构十分复杂,不同来源的HA的化学组成和结构差异较大,直接影响着HA与纳米材料在水环境中的环境行为和生态效应。在水相沉积物中的HA,氮和氢的含量较高,碳含量较低,且富含脂链;在泥炭和煤中,氮含量较低,碳含量较高;而土壤HA的碳和氮含量都较高且富含氧[13, 22]。研究表明不同来源的HA对于Fe3O4纳米颗粒悬浮性的影响具有较大差异,从而影响其在生态环境中的迁移、转化[23]。因此,不同来源的HA其自身所带官能团会影响其在TiO2 NPs表面的吸附以及在水环境中的悬浮/沉降性能。
在水环境中,不同来源的HA物理化学性质的差异会极大地影响其与纳米材料的相互作用,进而影响纳米材料以及其他化合物在水环境中的环境行为和生态效应。因此,研究不同水体理化要素条件下,不同来源HA在TiO2 NPs表面的吸附、悬浮/沉降行为及其机理对于全面掌握TiO2 NPs在水环境中的环境行为具有重要的意义。本文研究了在不同pH条件下,TiO2 NPs对两种不同来源的HA的吸附及其机理,以及吸附前后TiO2 NPs悬浮沉降性能的变化,为研究纳米材料在水环境中的环境行为及生态效应提供理论基础。
1 材料与方法 1.1 实验材料所有试剂均为分析纯,NaOH和HCl购于北京化工厂,TiO2 NPs购于百灵威科技有限公司(J & K Scientific,美国,Lot:CAS:13463-67-7),两种来源不同的腐植酸购于国际腐植酸协会(International Humic Substances Society,IHSS),分别来自于风化褐煤(Leonardite)提取的腐植酸(HAL,1S104H)以及水中(Suwannee River Ⅱ)提取的腐植酸(HAS,2S101H),两种不同来源腐植酸的核磁分析、元素组成及酸性官能团分析如表 1所示,所有试验用水由Millipore integral 5超纯水系统提供,出水电导率为18.2 MΩ·cm-1。
|
|
表 1 不同来源的HA的核磁、元素组成及酸性官能团分析 Table 1 NMR, elemental analysis and acidic functional groups analysis of different sources of HA |
分别用超纯水在1000 mL的容量瓶中溶解100 mg HAL和HAS样品,缓慢加入1 mol·L-1的NaOH溶液数滴,放入恒温摇床,温度为25 ℃、转速为120 r· min-1,经过24 h的振荡后进行定容,制备浓度为100 mg·L-1,并将HA储备液避光储存。通过稀释HA储备液配制浓度梯度为5、10、20、40、60 mg·L-1的HA溶液,使用安捷伦8453分光光度计,用1 cm光路长度的石英比色皿在波长为254 nm处进行紫外可见吸收光谱测量,从而得出HA浓度的标准曲线。
1.3 吸附平衡时间研究为了研究TiO2 NPs对于HA的吸附达到平衡所需要的时间,称取10 mg的TiO2 NPs加入到容量为100 mL的聚碳酸脂瓶中,然后加入40 mL超纯水,并用超声仪超声5 min使纳米颗粒在水溶液中溶解分散均匀,再加入10 mL HA储备液,最后将混合后的溶液pH调节至3.0±0.2(混合液中HA初始浓度为20 mg· L-1)。将样品放入摇床中以120 r·min-1的速度分别振荡(25 ℃)和平衡5、15、30 min和1、1.5、2、3、4、8、12、16、20、24 h。当悬浮液达到振荡平衡时间后,取上清液过0.45 μm的尼龙膜待测。采用254 nm波长的紫外可见吸收光谱对过滤后的溶液进行分析,HA浓度通过校准曲线测定。
为了定量描述两种HA在TiO2 NPs表面吸附随时间变化的情况,采用二级动力学方程进行拟合计算(公式1)。

|
(1) |
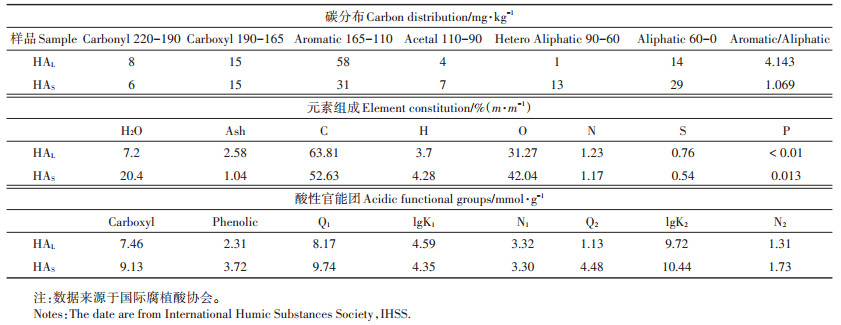
式中:k2是吸附速率常数,g·mg-1·min-1;qt是在任何时间被TiO2 NPs吸附的量,mg·g-1;qe是平衡时的吸附容量,mg·g-1。
初始吸附速率h0(mg·g-1·min-1)公式:
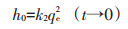
|
(2) |
将10 mg TiO2 NPs和10 mL HA储备液及40 mL超纯水加至聚碳酸脂瓶中,超声5 min(均同上述吸附时间研究),用0.1 mol·L-1的NaOH和HCl溶液调节悬浮液的pH为2.0到11.0,并放置在摇床上振荡平衡24 h(条件同上述吸附时间研究)。取上清液过0.45 μm过滤膜,样品在254 nm处进行紫外可见吸收光谱分析。
1.5 吸附等温线实验将10 mg TiO2 NPs加入到不同50 mL浓度HA溶液,HA浓度分别为5、10、15、20、30、40、50 mg·L-1,用0.1 mol·L-1的HCl溶液调节溶液pH为3.0。悬浮液放置在摇床上振荡平衡24 h(条件同上述吸附时间研究)。取上清液过0.45 μm过滤膜。样品在254 nm处进行紫外可见吸收光谱分析。通过TiO2 NPs吸附平衡前后吸光度的差异,确定HA的吸附量。
HA在TiO2 NPs表面的吸附等温线分别用Langmuir和Freundlich模型进行拟合。Langmuir主要用于理想状态的吸附模型,假设吸附反应是发生在均相表面上的单层吸附。而Freundlich是一种半经验模型,适用于低浓度下非均相表面的吸附[24]。
Langmuir方程为:
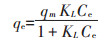
|
(3) |
式中:qm(mg·g-1)是最大吸附容量,qe(mg·g-1)是某一平衡浓度下单位二氧化钛纳米颗粒吸附HA的量,Ce(mg·L-1)是平衡时HA的浓度,KL是Langmuir吸附常数。
Freundlich方程为:

|
(4) |
式中:KF为Freundlich吸附系数;n为常数;Ce为吸附后HA的浓度,mg·L-1。
1.6 沉降实验pH对TiO2 NPs悬浮/沉降性能影响:将10 mg TiO2 NPs定容至50 mL,分别调节溶液pH为3、7、10,将混合溶液转移到1 cm石英比色皿中,置于紫外分光光度计,在508 nm下测定其动态吸光度Ce,其中初始吸光度为C0,以t为横坐标,Ce·C0-1的值为纵坐标做图,其中斜率即d(Ce·C0-1)·dt-1为沉降速率。
pH对TiO2 NPs悬浮/沉降性能影响:将10 mg TiO2 NPs分别加入10 mL HAL和HAS储备液,定容至50 mL,调节pH为3、7、10,待溶液达到相对应的平衡时间后(HAL:20 h,HAL:8 h),再将混合溶液转移到1 cm石英比色皿中,置于紫外分光光度计,在508 nm下测定其动态吸光度Ce,其中初始吸光度为C0,以t为横坐标,Ce·C0-1的值为纵坐标做图,其中斜率即d(Ce· C0-1)·dt-1为沉降速率。
1.7 表征透射电镜所用仪器为:H7500(TEM,日立,日本),电压为120 kV,电流30 mA。氮气吸附-解吸等温线测定-196用于Brunauer-Emmett-Teller(BET)测量(Micromeritics,美国),在测量之前,样品在50 ℃脱气,在相对压力(P·P0-1)0.06~0.20范围内计算BET比表面积。使用FT-IR光谱仪(Bruker,德国)测定了TiO2 NPs和不同来源的HA吸附前后的红外光谱(FT-IR),所有样品的测量均采用KBr球团法在室温下进行。X射线光电子能谱(XPS),采用英国Kratos公司Ultra DLD多功能光电子能谱仪对吸附HA前后TiO2 NPs进行XPS分析,激发源是AlKα(1 486.6 eV)X射线,并用污染碳的C 1s谱线(284.6 eV)校正。首先对试样进行全扫描,然后为确定各个元素的化合价合成键,对各个谱线进行扫描。数据采用Origin 8.0软件进行数据处理和制图。
2 结果和讨论 2.1 纳米二氧化钛表征由图 1a可以看到TiO2 NPs为粒径均匀的球形颗粒,粒径约为25~40 nm,且具有一定的分散性。图 1b为TiO2 NPs在溶液中随pH变化的电势变化。当溶液pH < 6.1时,TiO2 NPs表面带正电荷,且pH值越低电势越高。当pH不断升高时,TiO2 NPs表面正电荷数量逐渐减少,当pH升至6.1时,此时的Zeta电势为0,TiO2 NPs的等电点为pH=6.1。当pH值继续升高时,TiO2 NPs表面带负电荷并随着pH的不断增加,TiO2 NPs表面负电荷密度增加。

|
图 1 二氧化钛纳米颗粒透射电镜图(a)和TiO2 NPs的zeta电位在溶液中随pH变化(b) Figure 1 TEM image of TiO2 NPs(a)and zeta potential of TiO2 NPs in solution varying with pH(b) |
pH是水环境的重要理化参数,决定了金属氧化物纳米材料表面所带电荷的性质和密度。如图 2所示,两种HA在TiO2 NPs表面的吸附量随溶液pH的增加呈现下降趋势,在溶液pH=2.0时,HA的吸附量最大,分别为92.72 mg·g-1(HAL)、64.71 mg·g-1(HAS)。对于HAS,当溶液pH大于等电点时,就已经很难吸附到纳米颗粒表面,而对于HAL,在等电点之后还会有较小的吸附,直到pH大于10.0时,才很难吸附到纳米颗粒表面。当溶液pH小于TiO2 NPs等电点时,纳米颗粒表面带正电荷,带负电荷的HA能够通过静电引力作用吸附到TiO2 NPs表面,随着溶液pH值的不断升高,TiO2 NPs表面正电荷密度逐渐减少,吸附量也随之下降;当溶液pH大于等电点后,TiO2 NPs表面带负电荷,而同样带负电荷的HA则很难通过静电引力作用吸附到TiO2 NPs表面。当pH大于等电点时,HAL仍然会有少量吸附在纳米颗粒上。有研究表明疏水作用可促使水体中天然有机质中的疏水基团远离水分子,与水中的纳米材料发生吸附[25],而HAL含有更多的芳香族官能团,较HAS具有更强的疏水性,因此在pH大于等电点时,HAL的吸附量明显大于HAS很可能是由于疏水性作用的影响。因此,由于不同来源HA含有官能团种类和数量的差异,使其在TiO2 NPs表面吸附量随pH变化趋势也存在明显不同。

|
图 2 pH对TiO2 NPs吸附HA的影响 Figure 2 The effect of pH on adsorption of HA on TiO2 NPs surface |
图 3为不同平衡时间条件下,两种HA在TiO2 NPs表面的吸附量变化。由图 3a可见,在溶液pH值为3.0的条件下,HAL和HAS在TiO2 NPs表面的吸附量分别在20 h和8 h后基本未发生变化,达到吸附平衡。两种HA的吸附量随时间的变化趋势大致相同,但是两者的吸附量有着较大的差别,HAL的吸附量显著高于HAS,达到平衡时,HAL的吸附量55.02 mg·g-1显著高于HAS的吸附量32.85 mg·g-1。由于HA在TiO2 NPs表面的吸附机理主要是颗粒表面与HA组分基团之间的配体交换和静电引力起主要作用,且纳米颗粒吸附路径较短,因此TiO2 NPs对HA的吸附量在刚开始变化较大,而随着TiO2 NPs表面吸附点位慢慢减少至趋于饱和,HA的吸附量变化逐渐减小直到平衡。

|
图 3 HAL(a)和HAS(b)在TiO2 NPs表面的吸附平衡时间(溶液pH=3.0) Figure 3 Adsorption equilibrium time of HA on the surface of TiO2 NPs(Solution pH=3.0) |
HAL和HAS在TiO2 NPs表面的吸附等温线分别用Langmuir和Freundlich模型进行拟合。由公式(3)和(4)拟合得到两种HA在TiO2 NPs表面的Langmuir和Freundlich吸附等温线(图 4)。

|
图 4 HAL(a)和HAS(b)在TiO2 NPs表面的Langmuir和Freundlich吸附等温线 Figure 4 The Langmuir and Freundlich adsorption isotherm of HA on TiO2 NP surface |
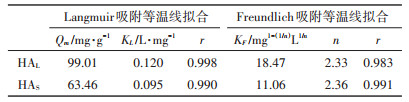
表 2为Langmuir和Freundlich吸附等温线拟合参数。HA在TiO2 NPs表面的吸附等温线均符合Langmuir和Freundlich方程,线性相关系数r > 0.95。Langmuir方程计算得到在25 ℃、pH=3.0条件下,HAL最大吸附量为99.01 mg·g-1,HAS最大吸附量为63.46 mg· g-1,吸附容量的差异主要是由于两种不同来源腐植酸的组分和结构的不同所造成的。Freundlich方程计算得到两种HA的吸附强度常数n为2.36和2.33,该常数说明在溶液pH=3.0时,TiO2 NPs对HA具有较强的吸附能力,主要原因是在pH=3.0时,TiO2 NPs表面带正电荷,带负电荷的HA可以通过静电引力作用吸附到TiO2 NPs表面。
|
|
表 2 HA在二氧化钛纳米颗粒表面Langmuir和Freundlich吸附等温线 Table 2 The Langmuir and Freundlich adsorption isotherm of HA on TiO2 NPs |
HAL和HAS在TiO2 NPs表面的吸附随时间变化的情况,采用二级动力学方程进行拟合计算(公式1),k2和h0可以通过t·qt-1和t作图得到的一条直线的斜率和截距计算得到,见图 5。

|
图 5 HA在TiO2 NPs表面吸附的二级方程吸附动力学拟合(溶液pH=3.0) Figure 5 Pseudo-second-order kinetic curves of adsorption of HA on the surface of TiO2 NPs(Solution pH=3.0) |
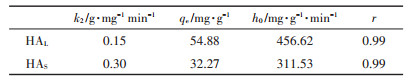
表 3为HA在TiO2 NPs表面吸附的二级吸附动力学拟合参数。其线性的相关系数r均大于0.99,说明两种HA在TiO2 NPs上的吸附动力学都满足拟二级动力学方程。由于两种HA组分和结构的不同,导致HAS在纳米颗粒上的吸附速率常数大概是HAL的2倍,但初始吸附速率HAL相比于HAS更高。
|
|
表 3 HA在TiO2 NPs表面的吸附动力学参数 Table 3 The adsorption kinetic model rate of adsorption of HA on the surface of TiO2 NPs |
不同pH值、离子强度和天然有机质存在条件下,纳米颗粒的悬浮/沉降行为有着较大的差异[17-18, 26-27]。在pH值较低时,纳米颗粒的Zeta电位一般为正,随着pH值的增加,其Zeta电位逐渐下降至负值。pH值与等电点的接近程度决定了纳米粒子之间的静电斥力,从而影响其悬浮/沉降行为。
如图 6所示,当pH为7.0时,其TiO2 NPs的沉降行为最为明显。而在pH为3.0和10.0条件下,TiO2 NPs则表现出稳定的悬浮状态。TiO2 NPs的等电点为6.1,当pH为7.0时,接近TiO2 NPs等电点,其表面不带电荷,颗粒间的经典排斥作用最小,TiO2 NPs更容易团聚而沉降。当pH为3.0和10.0时,距离等电点较远,TiO2 NPs表面电荷密度较大,颗粒之间的静电斥力导致其更易悬浮。

|
图 6 pH对TiO2 NPs悬浮/沉降行为的影响 Figure 6 Effect of pH on suspension/sedimentation behavior of TiO2 NPs |
图 7为HA对TiO2 NPs悬浮/沉降行为的影响。如图所示,在HA存在的条件下,TiO2 NPs能够在溶液中悬浮而不易沉降。当pH为3.0时,TiO2 NPs表面带有大量的正电荷,HA能够通过静电引力作用吸附到TiO2 NPs表面,HA可以增加纳米颗粒的净表面电荷,导致更高的静电斥力,颗粒间的净能量壁垒也会相应增强,进而导致团聚体破碎而分散悬浮[28-29]。同时,HA的吸附对TiO2 NPs有空间位阻作用,增强颗粒间的聚集能垒,使颗粒稳定悬浮在溶液中[30-31]。与未添加HA条件下相比较,当pH为7.0时,HA的吸附能够更为明显地促进TiO2 NPs的悬浮,主要是由于HA吸附到TiO2 NPs表面后不仅会增加表面电荷密度,还可能通过空间位阻作用阻碍颗粒间的团聚[19, 23],使得TiO2 NPs的悬浮性能增加。特别是HAL由于吸附量更大,其对TiO2 NPs的悬浮性能影响更加显著,这与前面不同pH条件下HAL和HAS的吸附量研究结果一致。对于不同来源的HA,HAL对TiO2 NPs的悬浮行为的影响相比于HAS更明显,主要是由于在相同pH条件下,HAL的吸附量高于HAS,静电排斥作用和空间位阻作用也更为明显。

|
图 7 HAL(a)、HAS(b)对TiO2 NPs悬浮/沉降行为的影响 Figure 7 Effect of HAL(a), HAS(b)on suspension/sedimentation behavior of TiO2 NPs |
图 8为TiO2 NPs表面吸附HAL和HAS前后傅里叶红外光谱的变化。通过比较TiO2 NPs吸附HAL和HAS前后的红外光谱图可以看到,在TiO2 NPs红外光谱图上3400 cm-1附近的特征峰为羟基组分,当HAL和HAS吸附到TiO2 NPs表面后,其羟基组分的特征峰出现在3420 cm-1附近,吸附后的峰强度相较于吸附前发生了明显的降低,证明了HAL和HAS都吸附到了TiO2 NPs表面,TiO2 NPs表面的羟基组分一方面与HA分子中的官能团相互作用而减少,另一方面由于HA分子量较大,吸附到TiO2 NPs表面后有包裹作用,因此减少了TiO2 NPs表面的羟基;HAL相比HAS含有更多的芳香族官能团,在TiO2 NPs+HAS的红外图谱中出现特征峰1387 cm-1,主要由HAL分子中的邻位取代芳香环伸展和羧酸组分的-OH弯曲振动产生;HAL分子中芳香族和羧酸中的C=C、COOH也在TiO2 NPs+HAL的红外图谱中有显示,而且特征峰位置也发生了变化,这与前面的研究结果类似[24]。同样在TiO2 NPs+HAS的红外图谱中,特征峰1720 cm-1为羧基中的C=O伸缩振动产生,相较于TiO2 NPs+HAL更为明显,主要是由于HAS分子中含有更多的羧酸组分。通过傅里叶红外光谱可以看到,特征峰1122~1127 cm-1在TiO2 NPs+ HAS图谱中更为明显,主要是由于HAS含有更多的脂肪族,而此处特征峰正是由于脂肪族中的C-O伸展产生;通过红外光谱图可以看到,HAL吸附到TiO2 NPs过程中,更多的芳香族官能团出现在TiO2 NPs+HAL的红外谱图中,而HAS则是脂肪族,而已有研究表明芳香族相比于脂肪族更容易促进二氧化钛纳米材料在水溶液中的悬浮稳定[32]。另外,HAL和HAS中其他的官能团,如亚甲基、甲基等都不同程度地出现在TiO2 NPs吸附HA后的红外光谱图中。通过比较TiO2 NPs吸附HAL和HAS前后傅里叶红外光谱图的变化,可以说明HAL和HAS都成功地吸附到TiO2 NPs表面,腐植酸分子中的官能团与TiO2 NPs表面的官能团发生了化学作用,除了HA由于分子量较大对TiO2 NPs有包裹作用以外,HAL和HAS中的芳香族和羧酸则能够与TiO2 NPs表面的羟基相互作用。

|
图 8 TiO2 NPs表面吸附HAL和HAS前后红外光谱变化 Figure 8 Infrared spectra of humic acid adsorbed on TiO2 NPs surface |
图 9为TiO2 NPs吸附HAL和HAS前后的XPS高分辨率谱图。由图中可以看到HAL和HAS吸附到TiO2 NPs表面后,XPS图谱中Ti 2p和O 1s特征峰的强度有明显的下降,主要原因是HA的吸附使其对TiO2 NPs具有包裹作用,XPS在研究过程中仅能穿透晶体表面约3~5 nm,所以TiO2 NPs表面的Ti和O元素的特征峰强度下降;同时,在HAL和HAS吸附到TiO2 NPs表面后,C 1s的特征峰强度明显上升,尤其是HAS,这主要是由于HA中的C元素显著高于TiO2 NPs表面,因此,在HAL和HAS吸附到TiO2 NPs表面后,C 1s特征峰强度明显上升。通过比较TiO2 NPs吸附HAL和HAS前后XPS高分辨率谱图的变化,可以说明HAL和HAS都成功地吸附到TiO2 NPs表面,HA由于分子量较大对TiO2 NPs具有一定的包裹作用。

|
图 9 HAL和HAS吸附到TiO2 NPs表面前后的XPS高分辨率谱图 Figure 9 XPS high-resolution spectrogram of HA adsorbed on TiO2 NPs surface |
通过不同来源HA对TiO2 NPs聚凝行为的影响研究,发现TiO2 NPs在水体环境中的吸附、悬浮/沉降行为极大地受到pH、HA来源等水环境理化要素的影响,因此TiO2 NPs进入不同水体环境后的迁移、转化、归趋及其潜在生态效应的差异性不容忽视。
(1)来自于风化褐煤(Leonardite)提取的腐植酸(HAL)和水中(Suwannee River Ⅱ)提取的腐植酸(HAS)在粒径约为25~40 nm的TiO2 NPs表面的吸附量差异较大。两种HA在TiO2 NPs表面的吸附量随溶液pH的增加呈现下降趋势,在溶液pH高于TiO2 NPs等电点后,HAL的吸附量明显大于HAS。HAL和HAS在TiO2 NPs表面的吸附等温线均符合Langmuir和Freundlich方程,以及在其表面吸附二级动力学方程的线性相关系数较高。
(2)溶液pH接近TiO2 NPs等电点时,TiO2 NPs表面电荷密度最低,TiO2 NPs更容易团聚而沉降。而HA的吸附能够促进TiO2 NPs在水溶液中的悬浮,尤其是在接近TiO2 NPs等电点时更为明显。相同pH条件下,吸附量更大的HAL对TiO2 NPs的悬浮行为的影响更加显著。
(3)通过比较TiO2 NPs吸附HAL和HAS前后傅里叶红外光谱图和X射线光电子能谱的变化,证明了HAL和HAS都成功地吸附到TiO2 NPs表面,腐植酸分子中的官能团与TiO2 NPs表面的官能团发生了化学作用,HA由于分子量较大对TiO2 NPs有包裹作用,HAL和HAS中的芳香族和羧酸能够与TiO2 NPs表面的羟基相互作用。
| [1] |
Chowdhury I, Cwierthy D M, Walker S L. Combined factors influencing the aggregation and deposition of nano-TiO2 in the presence of humic acid and bacteria[J]. Environmental Science & Technology, 2012, 46(13): 6968-6976. |
| [2] |
Kallio T, Alajoki S, Pore V, et al. Antifouling properties of TiO2:Photocatalytic decomposition and adhesion of fatty and rosin acids, sterols and lipophilic wood extractives[J]. Colloids and Surfaces A:Physicochemical and Engineering Aspects, 2006, 291(1/2/3): 162-176. |
| [3] |
Yu J G, Fan J J, Lv K L. Anatase TiO2 nanosheets with exposed(001) facets:Improved photoelectric conversion efficiency in dye-sensitized solar cells[J]. Nanoscale, 2010, 2(10): 2144. DOI:10.1039/c0nr00427h |
| [4] |
Han X G, Kuang Q, Jin M S, et al. Synthesis of titania nanosheets with a high percentage of exposed(001)facets and related photocatalytic properties[J]. Journal of the American Chemical Society, 2009, 131(9): 3152-3153. DOI:10.1021/ja8092373 |
| [5] |
Xiong L, Chen C, Chen Q, et al. Adsorption of Pb(Ⅱ)and Cd(Ⅱ)from aqueous solutions using titanate nanotubes prepared via hydrothermal method[J]. Journal of Hazardous Materials, 2011, 189(3): 741-748. DOI:10.1016/j.jhazmat.2011.03.006 |
| [6] |
Boxall A B, Tiede K, Chaudhry Q. Engineered nanomaterials in soils and water:How do they behave and could they pose a risk to human health?[J]. Nanomedicine, 2007, 2(6): 919-927. DOI:10.2217/17435889.2.6.919 |
| [7] |
Gondikas A, Kaegi R, Borovinskaya O, et al. Where is the nano? Analytical approaches for the detection and quantification of TiO2 engineered nanoparticles in surface waters[J]. Environmental Science:Nano, 2018, 5(2): 313-326. DOI:10.1039/C7EN00952F |
| [8] |
Liu W, Sun W L, Borthwick A G L, et al. Comparison on aggregation and sedimentation of titanium dioxide, titanate nanotubes and titanate nanotubes-TiO2:Influence of pH, ionic strength and natural organic matter[J]. Colloids and Surfaces A:Physicochemical and Engineering Aspects, 2013, 434: 319-328. DOI:10.1016/j.colsurfa.2013.05.010 |
| [9] |
Baggs R B, Ferin J, Oberdorster G. Regression of pulmonary lesions produced by inhaled titanium dioxide in rats[J]. Veterinary Pathology, 1997, 34(6): 592-597. DOI:10.1177/030098589703400607 |
| [10] |
Afaq F, Abidi P, Matin R, et al. Cytotoxicity, pro-oxidant effects and antioxidant depletion in rat lung alveolar macrophages exposed to ultrafine titanium dioxide[J]. Journal of Applied Toxicology, 1998, 18(5): 307-312. DOI:10.1002/(SICI)1099-1263(1998090)18:5<307::AID-JAT508>3.0.CO;2-K |
| [11] |
Long T C, Saleh N, Tilton R D, et al. Titanium dioxide(P25)produces reactive oxygen species in immortalized brain microglia(BV2):Implications for nanoparticle neurotoxicity[J]. Environmental Science & Technology, 2006, 40(14): 4346-4352. |
| [12] |
Adams L K, Lyon D Y, Alvarez P J J. Comparative eco-toxicity of nanoscale TiO2, SiO2, and ZnO water suspensions[J]. Water Research, 2006, 40(19): 3527-3532. DOI:10.1016/j.watres.2006.08.004 |
| [13] |
Aiken G R, Mcknight D M, Wershaw R L. Humic substances in soils, sediment, and water:Geochemistry, isolation, and characterization[M]. New York: John Wiley & Sons, 1985: 329-362.
|
| [14] |
Yang K, Lin D H, Xing B S. Interactions of humic acid with nanosized inorganic oxides[J]. Langmuir, 2009, 25(6): 3571-3576. DOI:10.1021/la803701b |
| [15] |
Wang X L, Lu J L, Xu M G, et al. Sorption of pyrene by regular and nanoscaled metal oxide particles:Influence of adsorbed organic matter[J]. Environmental Science & Technology, 2008, 42(19): 7267-7272. |
| [16] |
Kang S, Xing B S. Phenanthrene sorption to sequentially extracted soil humic acids and humins[J]. Environmental Science & Technology, 2005, 39(1): 134-140. |
| [17] |
Chen K L, Elimelech M. Influence of humic acid on the aggregation kinetics of fullerene(C60)nanoparticles in monovalent and divalent electrolyte solutions[J]. Journal of Colloid and Interface Science, 2007, 309(1): 126-134. DOI:10.1016/j.jcis.2007.01.074 |
| [18] |
Chen K L, Elimelech M. Interaction of fullerene(C60)nanoparticles with humic acid and alginate coated silica surfaces:Measurements, mechanisms, and environmental implications[J]. Environmental Science & Technology, 2008, 42(20): 7607-7614. |
| [19] |
Tang Z, Zhao X L, Zhao T H, et al. Magnetic nanoparticles interaction with humic acid:In the presence of surfactants[J]. Environmental Science & Technology, 2016, 50(16): 8640-8648. |
| [20] |
Yang S P, Bar-ilan O, Peterson R E, et al. Influence of humic acid on titanium dioxide nanoparticle toxicity to developing zebrafish[J]. Environmental Science & Technology, 2013, 47(9): 4718-4725. |
| [21] |
Lin D H, Ji J, Long Z F, et al. The influence of dissolved and surfacebound humic acid on the toxicity of TiO2 nanoparticles to Chlorella sp.[J]. Water Research, 2012, 46(14): 4477-4487. DOI:10.1016/j.watres.2012.05.035 |
| [22] |
Preston C M. Humus chemistry, genesis, composition and reactions[J]. Soil Science, 1995, 159(5): 356. DOI:10.1097/00010694-199505000-00012 |
| [23] |
汤智, 赵晓丽, 吴丰昌, 等. 不同来源腐植酸在纳米四氧化三铁上的吸附及对其沉降性的影响[J]. 环境化学, 2015, 34(8): 1520-1528. TANG Zhi, ZHAO Xiao-li, WU Feng-chang, et al. The interaction between Fe3O4 nanoparticle and different source humic acid, and the influence on nanoparticle suspension[J]. Environmental Chemistry, 2015, 34(8): 1520-1528. |
| [24] |
Kang S, Xing B S. Humic acid fractionation upon sequential adsorption onto goethite[J]. Langmuir, 2008, 24(6): 2525-2531. DOI:10.1021/la702914q |
| [25] |
Hur J, Schlautman M A. Molecular weight fractionation of humic substances by adsorption onto minerals[J]. Journal of Colloid and Interfaces Science, 2003, 264(2): 313-321. DOI:10.1016/S0021-9797(03)00444-2 |
| [26] |
Badawy A M E, Luxton T P, Silva R G, et al. Impact of environmental conditions(pH, ionic strength, and electrolyte type)on the surface charge and aggregation of silver nanoparticles suspensions[J]. Environmental Science & Technology, 2010, 44(4): 1260-1266. |
| [27] |
Loosli F, Le Coustumer P, Stoll S. Effect of natural organic matter on the disagglomeration of manufactured TiO2 nanoparticles[J]. Environmental Science:Nano, 2014, 1(2): 154-160. DOI:10.1039/c3en00061c |
| [28] |
Ghosh S, Mashayekhi H, Pan B, et al. Colloidal behavior of aluminum oxide nanoparticles As affected by pH and natural organic matter[J]. Langmuir, 2008, 24(21): 12385-12391. DOI:10.1021/la802015f |
| [29] |
Baalousha M. Aggregation and disaggregation of iron oxide nanoparticles:Influence of particle concentration, pH and natural organic matter[J]. Science of the Total Environment, 2009, 407(6): 2093-2101. DOI:10.1016/j.scitotenv.2008.11.022 |
| [30] |
Zhang Y, Chen Y S, Westerhoff P, et al. Impact of natural organic matter and divalent cations on the stability of aqueous nanoparticles[J]. Water Research, 2009, 43(17): 4249-4257. DOI:10.1016/j.watres.2009.06.005 |
| [31] |
Bian S W, Mudunkotuwa I A, Rupasinhge T, et al. Aggregation and dissolution of 4 nm ZnO nanoparticles in aqueous environments:Influence of pH, ionic strength, size, and adsorption of humic acid[J]. Langmuir, 2011, 27(10): 6059-6068. DOI:10.1021/la200570n |
| [32] |
Lin D H, Walker S L, Huang Q, et al. Influence of extracellular polymeric substances on the aggregation kinetics of TiO2 nanoparticles[J]. Water Research, 2016, 104: 381-388. DOI:10.1016/j.watres.2016.08.044 |