 2019, Vol. 38
2019, Vol. 38



微生物发酵床具有降解粪污的功效,其中的微生物起物质能量转化的作用,是发酵床良性运作的核心[1-2]。发酵床中的微生物种群丰富,肖荣凤等[3]自发酵床中分离了18种真菌,包含总状毛霉、尖孢枝孢菌、橘青霉和构巢曲霉等。刘国红等[4]研究表明发酵床中的芽孢杆菌含量多达4.41×108个·g-1,其中芽胞杆菌属、赖氨酸芽胞杆菌属和类芽胞杆菌属是优势菌。宦海林等[5]采用高通量测序的方法探寻了发酵床中的微生物组成,揭示发酵床的优势菌为拟杆菌门、厚壁菌门、变形菌门和放线菌门,包含节杆菌属、芽孢杆菌属、梭菌属、肠杆菌属、假单胞菌属、红球菌属等。笔者前期采用高通量测序发现发酵床中的主要细菌与宦海林等[5]的研究结果一致,同时发现发酵床中含有多种粪污降解菌,包括棒状杆菌属、芽孢杆菌属、枝芽孢杆菌属、假单胞菌属、放线菌属、乳杆菌属等[6]。
微生物发酵床垫料深度一般为60~100 cm,不同深度垫料的微环境有所差异,因此导致其中微生物种群和数量的差异。张学峰等[7]分离纯化了不同深度垫料的细菌,发现微生物发酵床30 cm处的垫料活菌数量大于表层垫料,推测微生物发酵床的核心发酵层是30 cm处的垫料层,此处的主要发酵菌株为地衣芽孢杆菌。而郑雪芳等[8]采用脂肪酸标记了不同深度垫料中的微生物,发现表层垫料(0~25 cm)中的微生物含量最高。由于培养条件限制,传统的分离培养法通常无法获得全部的环境微生物,因此可能会造成研究结果的差异。16S rDNA的高通量测序技术,无分离培养的过程,直接提取和克隆环境样品中所有微生物的DNA,获得环境中所有微生物的遗传信息,能够较为全面地揭示环境微生物的多样性。然而,基于宏基因分析养猪发酵床不同深度微生物组的研究未见报道,因此本研究采用高通量测序研究不同深度垫料的微生物群落,明确发酵床粪污降解的优势层,发掘潜在的降解菌,促进养殖废弃物的资源化利用。
1 材料与方法 1.1 试验材料微生物发酵床位于福建省福清渔溪现代设施农业样本工程示范基地,自2013年8月开始运行。发酵床垫料由70%椰糠和30%谷壳粉构成,垫料每年更换一次。采用五点取样法(以发酵床对角线的中点作为中心取样点,再在对角线上选择4个与中心取样点距离相等的点作为取样点,共计5处取样点),取10、11月和12月的不同深度垫料(表层10 cm、中层30 cm、深层50 cm),分别命名为OCT10cm、OCT30cm、OCT50cm、NOV10cm、NOV30cm、NOV50cm、DEC10 cm、DEC30cm、DEC50cm。
1.2 试验方法 1.2.1 微生物发酵床垫料总DNA的提取[9]每个样本称取500 mg垫料,按土壤DNA提取试剂盒FastDNA SPIN Kit for Soil的操作指南,分别进行总DNA的提取。调整终浓度至1 ng·μL-1开展后续试验。
1.2.2 微生物组16S rDNA V3~V4区测序采用原核生物16S rDNA基因V3~V4区通用引物338F(5′ -ACT CCT ACG GGA GGC AGC AG-3′)和806R(5′ - GGA CTA CHV GGG TWT CTA AT-3′,对各垫料样本总DNA进行PCR扩增(重复3次)。从3次重复中取相同体积混合后进行目的片段回收,所用胶回收试剂盒为AxyPrepDNA凝胶回收试剂盒(Axygen公司)。采用QuantiFluorTM-ST蓝色荧光定量系统(Promega公司)对回收产物进行定量检测。然后构建插入片段为350 bp的paired-end(PE)文库(TruSeqTM DNA Sample Prep Kit建库试剂盒,Illumina公司),经过Qubit定量和文库检测,HiSeq上机测序。
1.2.3 微生物组测序数据质控与分析对测序得到的原始数据进行拼接、过滤,得到有效数据。采用Mothur软件(version 1.36.1)基于有效数据进行OTUs(Operational Taxonomic Units)聚类和物种分类分析[10]。采用RDP classifier贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析[11]。从各个OTU中挑选出一条代表序列与已知物种的16S数据库(Silva,http://www.arb-silva.de)进行比对,完成物种注释;根据每个OTU中序列的条数,得到各个OTU的丰度值[12-13]。同时进行样品之间的共有及特有OTU数目分析、物种组成及物种差异性分析[14]、16S功能预测[15]。
相应的数据释放与SRA(Sequence Read Archive)数据库, 序列号分别为SRR5611224(OCT10cm)、SRR5611227(NOV10cm)、SRR5611230(DEC10cm)、SRR5611225(OCT30cm)、SRR5611228(NOV30cm)、SRR5611231(DEC30cm)、SRR5611226(OCT50cm)、SRR5611229(NOV50cm)和SRR5611232(DEC50cm)。
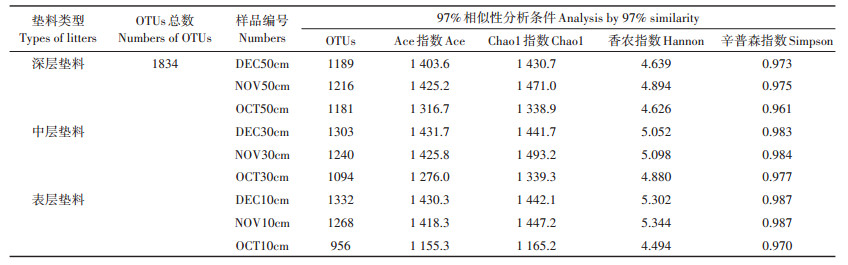
2 结果与分析 2.1 不同深度垫料细菌多样性测序分析经454焦磷酸测序,9个样本共获得1 045 225个有效序列,有效碱基数458 650 508 bp,序列平均长度438.8 bp,各样本分析结果见表 1。不同样品宏基因组分析结果存在差异:序列数量最大值为DEC10cm(140 350条),最小值为NOV50cm(101 537条)。表层、中层和深层垫料的短序列数分别为125 687、119 364和103 357。试验共检测到1834种OUT类型,包含32门、76纲、156目、303科、609属和939种细菌。其中,表层垫料包含31门、71纲、144目、283科、576属和891种细菌,中层垫料包含31门、72纲、146目、283科、580属和904种细菌,深层垫料包含30门、69纲、140目、268科、546属和853种细菌。
|
|
表 1 发酵床垫料微生物细菌群落宏基因组测序结果统计分析 Table 1 Similarity-based OTUs and species richness estimates of the bacterial phylotypes of the litters |
表层、中层和深层垫料OTUs种类分别为1701、1731种和1587种(图 1)。中层垫料OTUs种类最多,物种最为丰富,深层垫料细菌种类少。3组样本共有的OTU有1482种,表层10 cm样本与中层30 cm样本共有OTUs 1676种,10 cm与50 cm共有1513种OTUs,30 cm与50 cm样本共有155种OTUs。表层和中层样本共有OTUs种类最多,而表层和深层共有OTUs种类最少,表明相邻深度垫料趋向含有更多的共有OTUs。表层10 cm的样本,特有OTUs为26种,占表层总OTU的1.5%。中层30 cm样本特有OTUs为16种(0.9%),深层50 cm样本特有OTUs为14种(0.9%)。综上分析,微生物发酵床表层垫料具有更多的细菌和特有细菌种类,随着垫料深度增加,细菌种类减少;微生物发酵床相邻深度垫料具有更多的共有细菌种类。

|
图 1 样品韦恩图 Figure 1 Venn diagram denoting the unique and shared OTUs in the different samples |
微生物发酵床垫料共检测到32个细菌门(图 2)。在表层垫料中,占主要比例的细菌为拟杆菌门(25.3%)和变形菌门(25.9%);拟杆菌含量低于深层垫料同月份采集样本;放线菌门(10.2%)高于中层(7.6%)和深层垫料(5.4%)。在中层垫料中,含量高的细菌为拟杆菌门(27.8%)、变形菌门(25.1%)和厚壁菌门(17.0%)。在深层垫料中,主要的细菌门为拟杆菌门(33.3%)、变形菌门(13.0%)、厚壁菌门(15.2%)和糖杆菌门(14.7%),其中拟杆菌门含量高于表层和中层垫料。

|
图 2 微生物发酵床垫料在门水平的微生物组成 Figure 2 Relative abundances of predominant bacterial compositions on phylum levels |
微生物发酵床不同深度样本共获得303个细菌科(图 3)。在表层垫料中,黄杆菌科含量最高(7.4%)。在中层垫料中,黄单胞杆菌科(9.9%)、糖杆菌科(8.4%)和黄杆菌科(6.3%)是含量较高的细菌科。在深层垫料中,糖杆菌科(14.7%)含量最高,其次为螺旋体科(9.0%)、紫单胞菌科(7.6%)和海洋滑动菌科(7.1%)。微生物发酵床深层垫料不同月份样本具有相似的细菌群落构成,而表层和中层垫料不同月份样本的细菌群落结构差异很大。尤其是采集于10月份的微生物发酵床垫料,与相同深度不同月份的样本具有明显差异。10月份的表层和中层垫料中的主要细菌为糖杆菌门(17.7%)和变形菌门的黄单胞菌科(30.0%);11月份的表层和中层垫料中则是拟杆菌门的Cryomorphaceae(7.9%)和绿弯菌门的厌氧蝇菌科(8.7%)含量最高;12月份的表层和中层垫料中糖杆菌门(10.3%)和拟杆菌门的紫单胞杆菌科(9.6%)含量较高。

|
图 3 微生物发酵床垫料在科水平的微生物组成 Figure 3 Relative abundances of predominant bacterial compositions on family levels |
RDA结果显示(图 4),深度对垫料微生物多样性的影响大于月份对微生物群落的影响。厚壁菌门和绿弯菌门随着垫料深度的增加而增加,放线菌门、拟杆菌门和栖热菌门与垫料深度负相关。放线菌门在10月份的浅层垫料中含量最高,栖热菌门在12月份的表层垫料中含量最高。厚壁菌门、变形菌门、拟杆菌门和绿弯菌门与月份呈正相关,在11月份和12月份垫料中的含量高于10月份垫料。而放线菌门与月份呈负相关,在10月份垫料中含量最高。

|
图 4 垫料微生物与发酵床深度的RDA分析 Figure 4 Redundancy analysis(RDA)of the relationship between bacteria and depth |
综合分析,深层垫料有稳定的微生物群落,10、11月和12月采集的样本中,其主要优势菌都为糖杆菌门、螺旋体门和拟杆菌门。浅层垫料微生物群落变化较大,微生物分布与月份相关,如DEC30cm和DEC10cm(拟杆菌门、变形菌门和厚壁菌门),NOV30 cm和NOV10cm(拟杆菌门、变形菌门、厚壁菌门和绿弯菌门),OCT30cm和OCT10cm(拟杆菌门和变形菌门)有相似的微生物群落结构。发酵床养殖过程中,生物活动干扰表层和中层的垫料,因而导致发酵床表层垫料微生物群落的动态变化。
2.4 微生物发酵床不同深度垫料物种差异分析在门水平上,螺旋菌门在不同深度垫料间具有显著差异性(图 5),该门在深层垫料(9.2%)中含量远高于中层(2.7%)和浅层垫料(0.9%)。拟杆菌门随着垫料深度的增大而增多,在深层、中层和浅层垫料中的含量分别为33.3%、27.8%和25.3%。变形菌门则主要活跃在表层(25.9%)和中层垫料(25.1%)。放线菌门和异常球菌-栖热菌门在表层垫料中的含量大于中层和深层垫料,其中放线菌门在表层、中层和深层垫料中的含量分别为10.2%、7.6%和54%,异常球菌-栖热菌门在表层垫料中的含量为3.7%,是中层和深层垫料的1.8倍和3.0倍。随着深度的增加,底层垫料中的溶氧量降低,为厌氧菌提供了生长环境,因此在深层垫料中厌氧菌含量高。

|
图 5 不同深度垫料细菌在门水平上差异显著性分析 Figure 5 Significance difference of bacteria in litters of different depth on the phylunm level |
在科水平上,螺旋菌门的螺旋体科在深层垫料中的含量高于其他层垫料(图 6)。该菌是厌氧的腐生菌,可降解深层垫料中的有机质。拟杆菌门的海洋滑动菌科和拟杆菌目在深层垫料中的含量也高于浅层垫料,分别是表层垫料的4.0倍和19.3倍。而特吕珀菌科主要分布于表层(3.7%)和中层垫料(1.4%)中,在深层垫料(1.2%)中含量低。拟杆菌门的霜状菌在中层(3.7%)和表层(3.8%)垫料中含量高于深层垫料(0.5%)。

|
图 6 不同深度垫料细菌在科水平上差异显著性分析 Figure 6 Significance difference of bacteria in litters of different depth on the family level |
PICRUSt预测不同深度垫料中细菌的主要有机物降解途径的基因数目见图 7。表层垫料细菌群落中与氨基酸、碳水化合物和脂类代谢相关的基因拷贝数最多,深层最少,表明表层垫料中的细菌代谢最为旺盛,而深层垫料细菌的代谢较为缓慢。结合前文高通量测序的结果,表层垫料中细菌数量最多,提供了更多的代谢相关基因,因此代谢旺盛。如表层垫料中与氮素降解相关的硝酸还原酶在表层、中层和深层垫料的拷贝数分别为2928、1848和909,表层垫料中的含量高于深层垫料。因此,发酵床表层垫料中的细菌群落在养殖废弃物的降解中起主要作用。

|
图 7 不同发酵程度垫料中细菌群落氨基酸、碳水化合物和脂类代谢相关基因的分布 Figure 7 Functional prediction of bacterial community in amino acid, carbohydrate and lipid metabolism in litters of different fermentation grades |
本研究发现拟杆菌门、变形菌门和厚壁菌门是微生物发酵床的主要细菌。深层垫料与浅层垫料有着不同的微生物群落结构。发酵床垫料表层和中层中的细菌主要为异常球菌-栖热菌门的特吕珀菌科、变形菌门的黄单胞菌科和拟杆菌门的黄杆菌科细菌。特吕珀菌科细菌的最佳生长温度为50 ℃,可以抵御堆肥的高温,利用多种糖类、有机酸和氨基酸等有机质[16-17]。黄单胞菌科细菌在蚯蚓堆肥系统中起降解作用[18-19],还能够降解碳水化合物,是木质材料堆肥过程中的优势菌群[20-21],可能参与降解微生物发酵床垫料中的椰糠、谷壳以及猪粪中的碳水化合物。黄杆菌科细菌能够抵御抗生素胁迫,并能降解有机物,是堆肥中的常见菌[22];黄杆菌能够降解多糖,如纤维素、木聚糖、几丁质和葡聚糖[23-24];黄杆菌属的F. banpakuense可还原硝酸盐,参与堆肥中的氮循环[25]。
深层垫料中厌氧菌含量高,如螺旋体门的螺旋体科、拟杆菌门的腐螺旋菌科,微生物发酵床深层垫料缺氧环境适合其生长。螺旋体门的螺旋体科、拟杆菌门的紫单胞杆菌科和腐螺旋菌科、糖杆菌门、厚壁菌门的梭菌目是深层垫料中的主要有机质降解菌。紫单胞杆菌科在动物粪便降解中发挥作用[26-27];腐螺旋菌科在污泥处理中起重要作用[28-29]。糖杆菌目的细菌分布于土壤、沉积物、废水以及动物中,许多细菌能够厌氧发酵,分泌多种酶,参与有机物降解[30-31]。
在12月份的表层和中层垫料中,硫假单胞菌和寡源杆菌属含量较高,硫假单胞菌是厌氧菌,用硝酸盐氧化硫化物和乙酸盐,产生可利用的硫[32-33]。在11月份的表层和中层垫料中,厌氧蝇菌科含量较高,厌氧蝇菌科在降解碳水化合物的过程中有重要作用,Liang等[34]在长期发酵中发现厌氧蝇菌科在长链正烷烃生物降解的初始活化中起关键作用。在10月份表层和中层样本中,藤黄杆菌属含量最高,此属的某些细菌可分泌脂肪酶[35];某些细菌可降解甲胺磷,参与环境修复[36]。这些细菌可能参与了发酵床猪粪的降解过程。
此外,本研究首次在陆地生境中发现海洋细菌——霜状菌,霜状菌属于黄杆菌目,分离自海水、藻类等海洋生境[37-38]。本实验场所是一座围海造田后的农场,地处海边,推测霜状菌经由海水渗透和空气传播至猪场,这是首次在微生物发酵床中发现该类细菌,其功能及对发酵床的影响有待进一步研究。
4 结论(1)本研究采用高通量测序技术,分析了3种不同深度垫料中的细菌群落,结果发现表层垫料中微生物含量最高,表明发酵床表层是猪粪降解的主要功能层。
(2)发酵床表层和中层的优势菌为异常球菌-栖热菌门的特吕珀菌科、变形菌门的黄单胞菌科和拟杆菌门的黄杆菌科细菌;深层垫料中含有丰富的厌氧细菌,包括螺旋体科、腐螺旋菌科,参与发酵床中有机质的降解。
(3)微生物发酵床细菌资源丰富,蕴藏着许多木质素、多糖、蛋白质降解菌及未知功能的细菌,本研究为发酵床有机物降解及其微生物资源的利用提供了基础。
| [1] |
Chan D K O, Chaw D, Lo C Y. Development of an environmentally friendly and cost effective system for the treatment of waste in pig farming[J]. Journal of Agricultural Engineering Research, 1995, 56(3): 11-17. |
| [2] |
Deininger A, Tamm M, Krause R, et al. Penetration resistance and water-holding capacity of differently conditioned straw for deep litter housing systems[J]. Journal of Agricultural Engineering Research, 2000, 77(3): 335-342. |
| [3] |
肖荣凤, 刘波, 朱育菁, 等. 养猪微生物发酵床真菌空间分布特性研究[J]. 中国生态农业学报, 2018, 26(4): 493-504. XIAO Rong-feng, LIU Bo, ZHU Yu-jing, et al. Spatial distribution characteristics of fungal population in microbial fermentation bed for pig rearing[J]. Chinese Journal of Eco-Agriculture, 2018, 26(4): 493-504. |
| [4] |
刘国红, 刘波, 王阶平, 等. 养猪微生物发酵床芽胞杆菌空间分布多样性[J]. 生态学报, 2017, 37(20): 6914-6932. LIU Guo-hong, LIU Bo, WANG Jie-ping, et al. Spatial distribution pattern and community diversity of the Bacillus-like species in a fermentation bed system for pig rearing[J]. Acta Ecologica Sinica, 2017, 37(20): 6914-6932. |
| [5] |
宦海琳, 闫俊书, 周维仁, 等. 不同垫料组成对猪用发酵床细菌群落的影响[J]. 农业环境科学学报, 2014, 33(9): 1843-1848. HUAN Hai-lin, YAN Jun-shu, ZHOU Wei-ren, et al. Effects of different bedding litters on bacterial community in pig biobed[J]. Journal of Agro-Environment Science, 2014, 33(9): 1843-1848. |
| [6] |
Chen Q, Liu B, Wang J, et al. Diversity and dynamics of the bacterial community involved in pig manure biodegradation in a microbial fermentation bed system[J]. Annals of Microbiology, 2017, 67(7): 491-500. DOI:10.1007/s13213-017-1278-y |
| [7] |
张学峰, 周贤文, 陈群, 等. 不同深度垫料对养猪土著微生物发酵床稳定期微生物菌群的影响[J]. 中国兽医学报, 2013, 33(9): 1458-1462. ZHANG Xue-feng, ZHOU Xian-wen, CHEN Qun, et al. The influence of padding of different depth in native microorganism fermentation bed of pigs on microbial flora during steady period[J]. Chinese Journal of Veterinary Science, 2013, 33(9): 1458-1462. |
| [8] |
郑雪芳, 刘波, 朱育菁, 等. 磷脂脂肪酸生物标记法分析养猪发酵床微生物群落结构的空间分布[J]. 农业环境科学学报, 2018, 37(4): 804-812. ZHENG Xue-fang, LIU Bo, ZHU Yu-jing, et al. Spatial distribution of microbial communities in a fermentation bed based on phospholipid fatty acid biomarkers[J]. Journal of Agro-Environment Science, 2018, 37(4): 804-812. |
| [9] |
陈倩倩, 刘波, 王阶平, 等. 基于宏基因组方法分析养猪发酵床微生物组季节性变化[J]. 农业环境科学学报, 2018, 37(6): 1240-1247. CHEN Qian-qian, LIU Bo, WANG Jie-ping, et al. Seasonal variation, based on metagenomic analysis, in the bacterial community of a microbial fermentation-bed used for pig-raising[J]. Journal of Agro-Environment Science, 2018, 37(6): 1240-1247. |
| [10] |
Schloss P D, Westcott S L, Ryabin T, et al. Introducing mothur:Open-source, platform-independent, community supported software for describing and comparing microbial communities[J]. Applied Environmental Microbiology, 2009, 75(23): 7537-7541. DOI:10.1128/AEM.01541-09 |
| [11] |
Wang Q, Garrity G, Tiedje J, et al. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J]. Applied & Environmental Microbiology, 2007, 73(16): 5261-5267. |
| [12] |
Quast C, Pruesse E, Yilmaz P, et al. The SILVA ribosomal RNA gene database project:Improved data processing and web-based tools[J]. Nucleic Acids Research, 2013, 41: 590-596. |
| [13] |
Oberauner L, Zachow C, Lackner S, et al. The ignored diversity:Complex bacterial communities in intensive care units revealed by 16S pyrosequencing[J]. Scientific Reports, 2013, 3: 1413. DOI:10.1038/srep01413 |
| [14] |
Derrick E, Sebastian S, Janaki P, et al. Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen[J]. PLoS One, 2012, 7(11): e48289. DOI:10.1371/journal.pone.0048289 |
| [15] |
Langille M G I, Zaneveld J, Caporaso J G, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences[J]. Nature Biotechnology, 2013, 31(9): 814-821. DOI:10.1038/nbt.2676 |
| [16] |
Albuquerque L, Simões C, Nobre M F, et al. Truepera radiovictrix, gen. nov. sp. nov. a new radiation resistant species and the proposal of Trueperaceae. fam. nov.[J]. Fems Microbiology Letters, 2005, 247(2): 161-169. DOI:10.1016/j.femsle.2005.05.002 |
| [17] |
Ivanova N, Rohde C, Munk C, et al. Complete genome sequence of Truepera radiovictrix type strain(RQ-24T)[J]. Standards in Genomic Sciences, 2011, 4(1): 91-99. DOI:10.4056/sigs.1563919 |
| [18] |
Jin L, Kim K K, Im W T, et al. Aspromonas composti gen. nov. sp. nov. a novel member of the family Xanthomonadaceae[J]. International Journal of Systematic and Evolutionary Microbiology, 2007, 57(8): 1876-1880. DOI:10.1099/ijs.0.64472-0 |
| [19] |
Lin Z, Bai J, Zhen Z, et al. Enhancing pentachlorophenol degradation by vermicomposting associated bioremediation[J]. Ecological Engineering, 2016, 87(3): 288-294. |
| [20] |
Folman L, Gunnewiek P, Boddy L B W. Impact of white-rot fungi on numbers and community composition of bacteria colonizing beech wood from forest soil[J]. Fems Microbiology Ecology, 2008, 63(2): 181-191. DOI:10.1111/j.1574-6941.2007.00425.x |
| [21] |
Hervé V, Roux X L, Uroz S, et al. Diversity and structure of bacterial communities associated with Phanerochaete chrysosporium, during wood decay[J]. Environmental Microbiology, 2013, 16(7): 2238-2252. |
| [22] |
Bernardet J F, Segers P, Vanca M, et al. Cutting a gordian knot:Emended classification and description of the genus flavobacterium, emended description of the family flavobacteriaceae, and proposal of Flavobacterium hydatis nom. nov.(Basonym, Cytophaga aquatilis Strohl and Tait 1978)[J]. International Journal of Systematic Bacteriology, 1996, 46(1): 128-148. DOI:10.1099/00207713-46-1-128 |
| [23] |
Lee C C, Smith M, Kibblewhiteaccinelli R E, et al. Isolation and characterization of a cold-active xylanase enzyme from Flavobacterium sp.[J]. Current Microbiology, 2006, 52(2): 112-116. DOI:10.1007/s00284-005-4583-9 |
| [24] |
Rasmussen M A, Madsen S M, Stougaard P, et al. Flavobacterium sp. strain 4221 and Pedobacter sp. strain 4236 beta-1, 3-glucanases that are active at low temperatures[J]. Applied & Environmental Microbiology, 2008, 74(22): 7070-7072. |
| [25] |
Kim J J, Jin H M, Lee H J, et al. Flavobacterium banpakuense sp. nov. isolated from leaf-and-branch compost[J]. International Journal of Systematic and Evolutionary Microbiology, 2011, 61(Pt7): 1595-1600. |
| [26] |
Shkoporov A, Khokhlova E, Chaplin A, et al. Coprobacter fastidiosus gen. nov. sp. nov. a novel member of the family Porphyromonadaceae isolated from infant faeces[J]. International Journal of Systematic and Evolutionary Microbiology, 2013, 63(Pt11): 4181-4188. |
| [27] |
Sakamoto M, Lan P, Benno Y. Barnesiella viscericola gen. nov. sp. nov. a novel member of the family Porphyromonadaceae isolated from chicken caecum[J]. International Journal of Systematic and Evolutionary Microbiology, 2007, 57(Pt 2): 342-346. |
| [28] |
Paster B, Dewhirst F, Weisburg W, et al. Phylogenetic analysis of the spirochetes[J]. Journal of Bacteriology, 1991, 173(19): 6101-6109. DOI:10.1128/jb.173.19.6101-6109.1991 |
| [29] |
端正花, 潘留明, 陈晓欧, 等. 低温下活性污泥膨胀的微生物群落结构研究[J]. 环境科学, 2016, 37(3): 1070-1074. DUAN Zheng-hua, PAN Liu-ming, CHEN Xiao-ou, et al. Changes of microbial community structure in activated sludge bulking at low temperature[J]. Environmental Science, 2016, 37(3): 1070-1074. |
| [30] |
Tomonori K, ShiroY, Ryohei U, et al. Phylogenetic diversity and ecophysiology of Candidate phylum Saccharibacteria in activated sludge[J]. Fems Microbiology Ecology, 2016, 92(6): 78-89. |
| [31] |
Tadashi N, Speirs L, Junji F, et al. FISH probes targeting members of the phylum Candidatus Saccharibacteria falsely target Eikelboom type 1851 filaments and other Chloroflexi members[J]. Environmental Microbiology Reports, 2014, 6(6): 611-617. DOI:10.1111/1758-2229.12172 |
| [32] |
Tan W, Cong H, Chen C, et al. Bioaugmentation of activated sludge with elemental sulfur producing strain Thiopseudomonas denitrificans, X2 against nitrate shock load[J]. Bioresource Technology, 2016, 220: 647-650. DOI:10.1016/j.biortech.2016.08.093 |
| [33] |
Tan W, Jiang Z, Chen C, et al. Thiopseudomonas denitrificans gen. nov. sp. nov. isolated from anaerobic activated sludge[J]. International Journal of Systematic and Evolutionary Microbiology, 2015, 65(1): 225-238. |
| [34] |
Liang B, Wang L, Zhou Z, et al. High frequency of Thermodesulfovibrio spp. and Anaerolineaceae in association with Methanoculleus spp. in a long-term incubation of n-Alkanes-Degrading methanogenic enrichment culture[J]. Frontiers in Microbiology, 2016, 7: 1431-1444. |
| [35] |
Bresciani F, Santi L, Macedo A, et al. Production and activity of extracellular lipase from Luteibacter sp.[J]. Annals of Microbiology, 2014, 64(1): 251-258. DOI:10.1007/s13213-013-0657-2 |
| [36] |
Wang L, Wang G, Li S, et al. Luteibacter jiangsuensis sp. nov.:A methamidophos-degrading bacterium isolated from a methamidophos-manufacturing factory[J]. Current Microbiology, 2011, 62(1): 289-295. |
| [37] |
Lee D H, Choi E K, Moon S R, et al. Wandonia haliotis gen. nov. sp. nov. a marine bacterium of the family Cryomorphaceae, phylum Bacteroidetes[J]. International Journal of Systematic and Evolutionary Microbiology, 2010, 60(3): 510-514. DOI:10.1099/ijs.0.012674-0 |
| [38] |
Muramatsu Y, Takahashi M, Kamakura Y, et al. Salinirepens amamiensis gen. nov. sp. nov. a member of the family Cryomorphaceae isolated from seawater, and emended descriptions of the genera Fluviicola and Wandonia[J]. International Journal of Systematic and Evolutionary Microbiology, 2012, 62(9): 2235-2340. |