 2020, Vol. 39
2020, Vol. 39



我国每年有9.8亿t作物秸秆产生,其中有2.3亿t是蔬菜秸秆,蔬菜秸秆和其他作物秸秆最大的区别是含水量较高、易腐烂,快速资源化利用是解决问题的出路,既解决环境问题,又充分利用资源。废弃物肥料化技术已经成为农业废物资源化利用的重要技术之一[1]。国内大多数有机肥料产品达到无害化标准需发酵15~20 d,而优质有机肥料发酵一般需要45~ 60 d。腐质化周期长是堆肥技术主要瓶颈[1]。传统堆肥工艺周期长、肥效差,因此亟需缩短发酵周期、提高腐解品质。好氧发酵过程实质是大分子有机质在微生物作用下分解为小分子并形成腐殖质的过程。因此,研究快速好氧发酵过程中有机物料的微生物群落结构变化,对研究有机物料快速肥料化具有非常重要的指导意义。
近几年来关于有机物料肥料化过程中微生物群落变化的报道很多,主要是基于宏基因组学,通过高通量测序技术更精准、更大通量地解析堆肥生境中群落结构变化。2013年,Farage等[2]首次利用高通量测序技术分析堆肥生境微生物总DNA,获得3×106条序列,揭示变形菌门的乳酸杆菌在研究样本中的优势地位,从此拉开对堆肥生境中高效降解菌群及相关功能基因资源的新认识。López-gonzález等[3]发现堆肥过程中微生物群落多样性最高的阶段是中期。王秀红等[4]应用高通量测序技术,从门水平上解析了玉米条垛式好氧发酵过程起关键作用的细菌菌群。大量报道采用Illumina Miseq高通量测序研究废弃物堆肥微生物群落结构、多样性及其功能[5-6]。然而,宏基因组技术只能检测样本中种群的存在性,而宏转录组技术在研究原位微生物功能、发掘活性微生物资源方面具有天然的技术优势。
宏转录组以研究复杂环境原位功能微生物总RNA为研究对象,因此表征的特定微生物处于生理活性状态。因此,与宏基因组相比较,宏转录组具有原位研究的优势[7]。宏转录组分析采集环境样品,提取总RNA,反转录为cDNA,然后借助二代测序对cDNA进行测序。近年来宏转录组学广泛应用到海洋和土壤等生境研究[8],但应用宏转录组技术分析有机物料好氧发酵的微生物群落变化的报道鲜见。本研究以蔬菜秸秆快速腐熟样品为研究对象,借助宏转录组技术,分析活性微生物群落结构变化,旨在更精准地分析好氧发酵过程中微生物种群结构变化,为解析发酵过程中起关键作用的微生物菌群、进一步改善发酵工艺、提高腐殖化效率提供指导依据。
1 材料与方法 1.1 发酵原料收集北京市农林科学院院内温室种植的西红柿秸秆为原料,前期收集的西红柿秸秆晒到半干状态,再收集新鲜的西红柿秸秆,两者混合一起粉碎至3~5 cm,干湿西红柿秸秆混合物含水量72.41%。发酵用牛粪取自北京昌平区中国农机院生态农业科技园养牛场,牛粪为半风干状态,含水量50.23%。
1.2 发酵方法有机废弃物原料按照一定配比充分混合,C/N在25~30,含水量控制在55%~60%。借助卧式生物好氧反应设备进行发酵(哈尔滨华美亿丰复合材料有限公司),设备容积6 m3,装料量4 m3左右。设备参数设置:温度低于40 ℃时,每12 h通风补氧20 min;40~ 50 ℃时,每8 h通风补氧20 min;当50~69 ℃时,每6 h通风补氧20 min;当温度高于69 ℃,持续通风补氧。参照文献Zhu等[9]和Saludes等[10]把发酵进程划分为4个不同时期(升温期、高温期、降温期、腐熟期,在本文中分别缩写为SW、GW、JW、HS)进行取样,取样前转动罐体5 min,从罐体前、中、后3个位置取样混合均匀;再次转动5 min后,再次从3个位置取样;如此反复取3次样品。每个时期取3个平行样品,共12个样品。样品一部分测定理化指标和用于发芽试验,另一部分-80 ℃保存,待分子生物学分析。
1.3 检测指标与方法按照有机物料与水1:10的比例混合,1 h振荡并静止后,检测pH值。干燥失重法测定含水量。罐体内3个不同部位设有3个温度计,每隔1 h采集温度一次。采用重铬酸钾容量法-外加热法测定有机质,凯氏定氮法测定全氮[11]。速效磷采用钼锑抗比色法测定[12]。铵态氮测定使用靛酚蓝比色法;硝态氮测定采用紫外分光光度法[13]。发芽指数(Germination index,GI)测定参照宋春丽[11]的方法。
1.4 样品细菌群落高通量测序和定量PCR分析使用PowerSoil™ Total RNA Isolation Kit(MoBio)试剂盒,提取样品总RNA,每个样品取2 g用于RNA提取试验,按试剂盒说明书进行操作。上述样品转录组总RNA反转录后的cDNA作为模板,利用微生物16S rRNA基因V3~V4区通用引物338F/806R[14-15](引物序列338F:5′ - ACTCCTACGGGAGGCAGCA-3′;806R:5′ - GGACTACHVGGGTWTCTAAT-3′)进行PCR扩增。PCR反应条件如下:94 ℃ 5 min;94 ℃ 30 s,55 ℃ 30 s,72 ℃ 60 s,30个循环;72 ℃ 7 min。PCR产物通过琼脂糖凝胶电泳和NanoDrop分析确保样品总RNA无污染。取RNA样品使用试剂1st Strand cDNA Synthesis Kit(TaKaRa)反转录合成cDNA。将反转录合成的cDNA样品送至北京奥维森基因科技有限公司应用Illumina MiSeq平台测序。
使用SYBR Green定量PCR法测定16S rRNA基因,反应在ABI 7500Real-time PCR(ABI,USA)仪器上进行,具体方法参照文献[16]。
1.5 数据分析利用QIIME软件对原始序列经过去除引物接头、序列拼接、指控过滤等分析处理之后得到有效数据。所有高质量序列在97%相似度水平进行分析,得到不同操作分类单位(Operational taxonomic unit,OTU)。参照Greengenes数据库,对OUT进行注释。应用Mothur软件分析样品细菌Alpha多样性指数。
采用Microsoft Excel 2010进行数据整理,使用SPSS 21.0统计分析软件进行数据分析,使用CANOCO 5.0对理化因子和细菌群落组成进行典范对应分析(CCA)。
2 结果与分析 2.1 不同发酵时期温度变化、理化性状与发芽指数分析好氧发酵过程中,堆体发生复杂的生化反应产生自热效应,温度升高越快,说明微生物活性越强,有机质降解的速度越快。堆温取决于环境温度、堆体初始温度和微生物活性。升温期又叫中温期,嗜温微生物活跃;本研究工艺条件下,50 h左右进入高温期,嗜热微生物主导;温度升至70 ℃以上时,绝大多数嗜热微生物失活,有机物降解缓慢,开始降温。蔬菜秸秆好氧发酵不同时期温度变化曲线见图 1,进罐时物料平均温度为30 ℃,第1次取样温度在45 ℃左右;温度到达55 ℃之后至最高温期间进行第2次取样;第3次取样是降温期,温度在45~50 ℃;温度降到40 ℃左右,趋于稳定后进行第4次取样。

|
图 1 好氧发酵温度变化曲线图 Figure 1 Curve of aerobic fermentation temperature change |
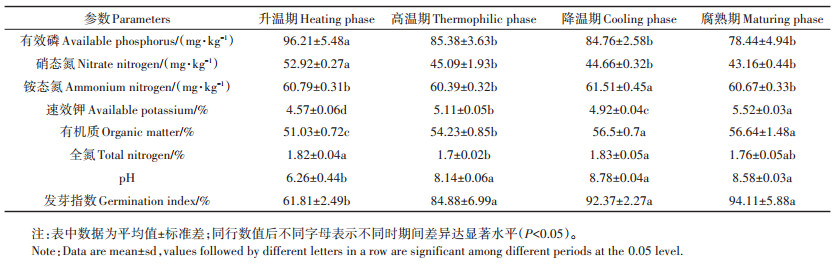
发酵体系内理化因子随着发酵进程的推进发生了差异性变化(见表 1),如有效磷和硝态氮,除了升温期较高,其余3个时期未发生显著变化;速效钾除了降温期,总体呈现增加趋势。pH值是影响微观环境微生物活性的重要环境因素之一,堆肥最适pH为6.5~8.5;高温期pH在7.5~8.5范围时,微生物分解能力最强;腐熟时pH一般呈弱碱性,在8~9左右。本研究好氧发酵蔬菜秸秆的升温期pH值为6.26,腐熟时pH为8.58(表 1),符合腐熟堆肥的一般要求。pH基本呈现从低到高的变化趋势,可能是因为升温期微生物反应剧烈,产生了大量有机酸,但随着试验条件下通氧充分,有机酸挥发使得pH值逐渐上升;另外,高温期之后,可能由于物料中微生物分解半纤维素、纤维素和蛋白质等有机物形成了弱碱性物质,导致pH上升。发芽指数值能有效表征种子生根对堆肥有机物料的植物毒性物质的响应,能够反映堆肥腐熟程度,也是较为可靠的生物学指标。发芽指数(发酵初期为61.81%)随着发酵时间的增加而增加,腐熟阶段达到94.11%,说明快速好氧发酵蔬菜秸秆废弃物达到了无害化。
|
|
表 1 不同发酵时期物料理化因子和发芽指数 Table 1 Physicochemical factors and GI of organic materials during the different fermentation periods |
蔬菜秸秆快速发酵不同发酵时期的有机物料样品16S rRNA基因丰度测定结果如图 2所示,基因拷贝数在(2.16~9.88)×1011 copies·g-1之间变化,高温期的基因丰度最高(9.88×1011 copies·g-1),升温期的最低,高温期比升温期高出7.72×1011 copies·g-1。而降温期和腐熟期差异不显著,分别是3.12×1011 copies· g-1和3.28×1011 copies·g-1。高温期的基因丰度是升温期的4.57倍,是降温期的3.17倍。

|
图 2 不同发酵时期16S rRNA基因丰度拷贝数 Figure 2 Copy numbers of 16S rRNA gene during the different fermentation periods detected by real-time PCR |
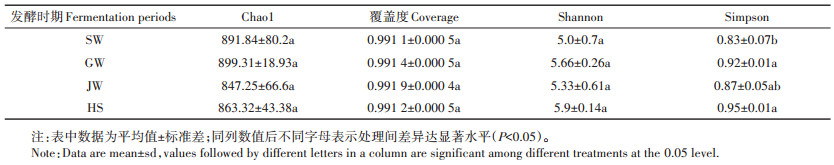
高通量Illumina MiseqTM测序16S rRNA基因分析,经过片段拼接和质控过滤后,获得蔬菜秸秆发酵4个时期样品的有效序列覆盖度均达到99%以上(表 2),说明本研究的测序深度可用于描述整个蔬菜秸秆废弃物快速好氧发酵条件下,处于生理活性状态的细菌群落结构变化情况。表 2为基于97%的相似水平分析有效序列的多样性,不同时期的Chao1指数和Shannon指数没有显著性差异。只有Simpson指数不同处理之间存在显著差异,升温期Simpson指数最低(0.83),即多样性最高,降温期(0.87)次之;高温期(0.92)和腐熟期(0.95)多样性相对较低。
|
|
表 2 不同发酵时期微生物多样性指数变化分析 Table 2 Microbial diversity during the different fermentation periods |
在蔬菜秸秆快速好氧发酵的4个时期,菌群门类随着发酵时间变化逐渐发生演替变化。在4个时期,主要优势菌门均是厚壁菌门(Firmicutes),但是相对丰度变化很明显,升温期、高温期、降温期、腐熟期的相对丰度分别是61.65%、94.37%、95.16%、89.23%。升温期的放线菌门(Actinobacteria)相对丰度高于变形菌门(Proteobacteria);但高温期和降温期正好相反;而腐熟期的第2高菌门和第3高菌门分别是Proteobacteria和拟杆菌门(Bacteroidetes)。软壁菌门(Tenericutes)虽不是优势菌门,但随着发酵进程的推进而发生了显著的变化(图 3),从升温期的0.02%,逐渐升高到0.21%。

|
图 3 不同发酵时期细菌群落优势菌门及相对丰度 Figure 3 Bacterial community composition and relative abundance of dominant phylum during different fermention periods |
整个发酵期间共检测到16个菌门,76个目,221个菌属,挑选4个不同时期代表性细菌菌属74个,相对丰度分析如图 4所示。不同发酵时期都存在的菌属45个,而主要菌属(至少在3个时期相对丰度大于0.1%的)有13个,主要是海洋芽孢杆菌属(Oceanobacillus)、乔治菌属(Georgenia)、高温双岐菌属(Thermobifida),Sinibacillus、嗜盐菌属(Halocella)、Vulgatibacter,Caldicoprobacter、芽孢杆菌属(Bacillus)等。在升温期优势菌属是Oceanobacillus和Gracilibacillus,相对丰度均大于18%;在高温期与降温期优势菌属是Oceanobacillus和Sinibacillus,相对丰度均大于7%;而腐熟期Sinibacillus的相对丰度最高(大于30%以上)。升温期相对丰度较高,且存在其他时期极低或未检出的菌属,如Salinimicrobium、Enteractinococcus、Glycomyces、Truepera。Sinibacillus随着发酵时间推移发生显著差异变化,相对丰度在升温期、高温期、降温期、腐熟期分别是3.45%、7.31%、9.79%、31.89%。Bacillus在升温期仅有0.91%,而在高温期和降温期均达4%以上,腐熟期又降到2.43%。Caldicoprobacter在升温期和高温期均低于0.8%,而降温期升高到1.56%,腐熟期降到1.07%。Moheibacter在前3个时期均低于0.3%,但腐熟期丰度达到1.2%以上,是增幅较大的菌属。

|
图 4 不同发酵时期物料细菌群落主要菌属组成及相对丰度 Figure 4 Bacterial community composition and relative abundance at genus level during different fermentation periods |
通过CCA分析不同时期样品细菌群落结构与理化因子之间的关系(图 5),发现高温期与降温期的群落结构最相似,升温期与其他几个时期的群落结构相距最远。参与分析的环境因子合计解释量为90.0%,说明理化因素显著影响好氧发酵过程中细菌的群落结构,Axis 1和Axis 2排序轴解释量分别为57.14%和23.30%。各个因子影响顺序:pH>速效钾>有机质>硝态氮>总氮>有效磷>铵态氮,其中pH解释量为54.0%(P=0.006),速效钾解释量为17.9%(P=0.004)。由此可知,发酵体系中的pH变化是影响体系内微生物群落结构的重要因素,也是影响发酵速率的关键因素之一,在发酵工艺过程中调控发酵体系pH,可能是提高发酵速率的一个有效途径。

|
图 5 不同发酵时期微生物群落与理化因子的CCA分析 Figure 5 CCA analysis between physic-chemical parameters and microorganisms |
通过定量PCR技术分析16S rRNA基因丰度,可知不同时期细菌丰度存在显著差异,高温期细菌丰度显著高于其他时期,是升温期的4.6倍。研究结果表明随着温度的升高,细菌生长能力增强,在高温期达到峰值;随着发酵进程的推进,部分嗜温细菌将会死亡,所以降温期细菌丰度明显低于高温期;而降温期和腐熟期的细菌丰度相对稳定。Tang等[17]借助醌类图谱法研究牛粪和稻草堆肥时也发现发酵后期细菌数明显低于高温期的现象。微生物数量的变化可以间接地表征堆肥腐熟度,可作为参考指标[18],本研究发酵过程中微生物数量经历了从低到高,再由高降低,最后趋于稳定的变化过程,对应的有机物料在微生物作用下,经历了一个快速腐解的变化过程。
3.2 不同发酵时期细菌群落多样性分析与宏基因组相比,宏转录组具有原位研究的优势[7],本研究基于宏转录组分析,挖掘原位活性微生物。细菌多样性变化的分析结果表明,Chao1指数、Shannon指数不同温度时期差异性不大,这与滑留帅等[19]在研究牛粪发酵过程中细菌多样性分析结果一致。而Simpson指数分析发现升温期的多样性最高,随着温度升高多样性降低,当高温期到降温期时,细菌多样性再次升高,到腐熟阶段多样性逐渐降低。徐小楠[20]和郭亚萍等[21]研究发现堆肥不同时期微生物的多样性总体趋势是从高到低,腐熟阶段的多样性指数最低,这与本研究结果(总体趋势是高-低-高-最低)基本一致,活性细菌菌群多样性变化结果显示,随着温度升高只有耐高温细菌有活性,所以高温阶段活性细菌多样性指数有所下降,宏转录组分析相对传统方法研究结果更有针对性,温度的变化促使活性细菌菌群多样性在不断演变。
3.3 不同发酵时期细菌群落结构演替分析不同发酵时期,有机物料中细菌群落结构发生了巨大的变化,不同时期的相对丰度最高的菌门均为Firmicutes,但丰度值差异很大,这一发现与牛粪秸秆堆肥体系中应用高通量测序技术检测到升温期和高温期最高优势菌门均为Proteobacteria的结果不同[22],这种不同可能是分析手段不同所致,报道是基于DNA水平分析,而本研究方法基于RNA水平。此外,Firmicutes更适合高温,在快速发酵体系内,代谢活性较强,在纤维素降解过程中发挥着重要作用[23]。Proteobacteria是各时期优势菌门,但在各时期的地位存在一定差异,这个发现与王秀红等[4]研究结果相同。Bacteroidetes在4个阶段,经历了升高到减少、再升高的变化,在腐熟阶段成为优势菌门。Ma等[24]报道在猪粪和小麦秸秆堆肥发酵过程中Firmicutes、Proteobacteria、Bacteroidetes、Actinobacteria、Tenericutes和芽单胞菌门(Gemmatimonadetes)6个优势菌门,在本研究中Gemmatimonadetes不是优势菌门,可能与发酵的原料不同有关系,也可能是因为宏基因组检测到的优势菌门在活性细菌中并非优势菌门。非优势菌门在数量上也发生着成倍数的增减变化,比如Tenericutes的相对丰度在腐熟期是高温期的10.5倍,说明菌群结构随着发酵进程推进发生着群落演替。基于属水平分析发现菌属类别和相对丰度也发生了显著差异变化。研究发现在蔬菜秸秆快速好氧发酵过程中,Oceanobacillus是优势菌属,前人研究发现Oceanobacillus具有分泌嗜碱胞外酶和特殊活性脂肪酸的潜力[25],蔬菜秸秆有机物料腐解过程pH分析结果表明,发酵体系处于Oceanobacillus菌属适合生长的弱碱性环境,利于此菌属发挥分泌特殊的有机酸和酶类的作用,因此可能加速发酵进程和提高腐殖化程度,但具体还需要进一步研究验证。以后可尝试把此菌属作为腐熟剂接种到有机物料堆肥体系中。通过对不同发酵时期的菌群结构分析,为提高发酵速率和有机物料腐熟品质方面的研究提供理论依据。
4 结论通过对蔬菜秸秆快速好氧发酵过程中活性细菌菌群丰度、多样性、结构等进行分析,可知菌群数量、多样性、组成和相对丰度均有显著变化。细菌数量在高温期最高,升温期最低;细菌多样性除Simpson指数外,其余指数总体差异性不显著;不同时期丰度最高的菌门均为Firmicutes,但相对丰度差异显著;不同时期菌属组成和相对丰度之间有明显差异,升温期存在其他时期检测不到的菌属;通过CCA分析可知pH是影响微生物群落结构的主要因子。
| [1] |
Lim S L, Leel H, Wu T Y. Sustainability of using composting and vermicomposting technologies for organic solid waste biotransformation:Recent overview, greenhouse gases emissions and economic analysis[J]. Journal of Cleaner Production, 2016, 111: 262-278. DOI:10.1016/j.jclepro.2015.08.083 |
| [2] |
Farage M L, Principal A L, Pascon R C, et al. Metagenomic analysis of a tropical composting operation at the São Paulo Zoo Park reveals diversity of biomass degradation functions and organisms[J]. PLoS One, 2013, 8(4): e61928. DOI:10.1371/journal.pone.0061928 |
| [3] |
López-gonzález J A, Suárez-estrella F, Vargas-garcía M C, et al. Dynamics of bacterial microbiota during lignocellulosic waste composting:Studies upon its structure, functionality and biodiversity[J]. Bioresource Technology, 2015, 175: 406-416. DOI:10.1016/j.biortech.2014.10.123 |
| [4] |
王秀红, 李欣欣, 史向远, 等. 好氧堆肥微生物代谢多样性及其细菌群落结构[J]. 环境科学研究, 2018, 31(8): 1457-1463. WANG Xiu-hong, LI Xin-xin, SHI Xiang-yuan, et al. Microbial metabolism diversity and bacterial flora structure during aerobic composting[J]. Research of Environmental Sciences, 2018, 31(8): 1457-1463. |
| [5] |
Khalld A, Rajandas H, Parimannan S, et al. Insights into microbial community structure and diversity in oil palm waste compost[J]. 3Biotech, 2019, 9: 364. |
| [6] |
Zhu L J, Zhao Y, Zhang W S, et al. Roles of bacterial community in the transformation of organic nitrogen toward enhanced bioavailability during composting with different wastes[J]. Bioresource Technology, 2019, 285: 121326. DOI:10.1016/j.biortech.2019.121326 |
| [7] |
赵俊, 莫永亮, 贾仲君. 土壤宏转录组RNA的提取方法评价[J]. 微生物学报, 2018, 58(4): 724-743. ZHAO Jun, MO Yong-liang, JIA Zhong-jun. Assessment of methodspecific bias associated with RNA extractions for metatranscriptomics in three geographically distinct paddy soils with different origin of parent materials[J]. Acta Microbiologica Sinica, 2018, 58(4): 724-743. |
| [8] |
马海霞, 张丽丽, 孙晓萌, 等. 基于宏组学方法认识微生物群落及其功能[J]. 微生物学通报, 2015(5): 102-112. MA Hai-xia, ZHANG Li-li, SUN Xiao-meng, et al. Understanding microbial communities and their functions by meta-omics approaches[J]. Microbiology China, 2015(5): 102-112. |
| [9] |
Zhu L J, Zhao Y, Zhang W S, et al. Roles of bacterial community in the transformation of organic nitrogen toward enhanced bioavailability during composting with different wastes[J]. Bioresource Technology, 2019, 285: 121326. DOI:10.1016/j.biortech.2019.121326 |
| [10] |
Saludes R B, Iwabuchi K, Miyatake F, et al. Characterization of dairy cattle manure/wallboard paper compost mixture[J]. Bioresource Technology, 2008, 99(15): 7285-7290. DOI:10.1016/j.biortech.2007.12.080 |
| [11] |
宋春丽. 蔬菜废弃物堆肥化技术研究[J]. 农学学报, 2019, 9(9): 40-44. SONG Chun-li. Study on composting techniques of vegetable wastes[J]. Journal of Agriculture, 2019, 9(9): 40-44. |
| [12] |
李帆, 钱坤, 武际, 等. 过磷酸钙用量对猪粪堆肥过程及磷形态变化的影响[J]. 植物营养与肥料学报, 2017, 23(4): 1037-1044. LI Fan, QIAN Kun, WU Ji, et al. Influence of applying calcium superphosphate on swine manure composting and phosphorus transformation[J]. Journal of Plant Nutrition and Fertilizers, 2017, 23(4): 1037-1044. |
| [13] |
李帆, 王静, 武际, 等. 尿素硝酸铵调节碳氮比促进小麦秸秆堆肥腐熟[J]. 植物营养与肥料学报, 2019, 25(5): 832-840. LI Fan, WANG Jing, WU Ji, et al. Fast production of wheat straw aerobic compost through regulating C/N ratio with urea ammonium nitrate solution[J]. Journal of Plant Nutrition and Fertilizers, 2019, 25(5): 832-840. |
| [14] |
Hong C, Si Y X, Xing Y, et al. Illumina MiSeq sequencing investigation on the contrasting soil bacterial community structures in different iron mining areas[J]. Environmental Science and Pollution Research, 2015, 22(14): 10788-10799. DOI:10.1007/s11356-015-4186-3 |
| [15] |
Ke W, Chu C, Li X K, et al. Succession of bacterial community function in cow manure composing[J]. Bioresource Technology, 2018, 267: 63-70. DOI:10.1016/j.biortech.2018.06.028 |
| [16] |
丁建莉, 姜昕, 关大伟, 等. 东北黑土微生物群落对长期施肥及作物的响应[J]. 中国农业科学, 2016, 49(22): 4408-4418. DING Jian-li, JIANG Xin, GUAN Da-wei, et al. Responses of micropopulation in black soil of northeast China to long-term fertilization and crops[J]. Scientia Agricultura Sinica, 2016, 49(22): 4408-4418. |
| [17] |
Tang J C, Kanamori T, Inoue Y, et al. Changes in the microbial community structure during thermophilic composting of manure as detected by the quinone profile method[J]. Process Biochemistry, 2004, 39(12): 1999-2006. DOI:10.1016/j.procbio.2003.09.029 |
| [18] |
弓凤莲, 杨义, 于淑婷, 等. 市政污泥堆肥过程参数变化及腐熟度综合评价[J]. 中国给水排水, 2014(21): 128-131. GONG Feng-lian, YANG Yi, YU Shu-ting, et al. Parameter change and comprehensive evaluation of maturity during municipal sewage sludge composting process[J]. China Water & Wastewater, 2014(21): 128-131. |
| [19] |
滑留帅, 王璟, 徐照学, 等. 16S rRNA基因高通量测序分析牛粪发酵细菌多样性[J]. 农业工程学报, 2016, 32(增刊2): 311-315. HUA Liu-shuai, WANG Jing, XU Zhao-xue, et al. Analysis of bacterial diversity during cattle manure fermentation with 16S rRNA gene high-throughput sequencing[J]. Transactions of the Chinese Society of Agricultural Engineering, 2016, 32(Suppl 2): 311-315. |
| [20] |
徐小楠.不同物料堆肥细菌多样性分异评价[D].哈尔滨: 东北农业大学, 2014. XU Xiao -nan. Analysisy of bacterial diversity in the law of different materials compost[D]. Harbin: Northeast Agricultural University, 2014. |
| [21] |
郭亚萍, 张国庆, 陈青君, 等. 双孢蘑菇堆肥过程中细菌群落结构分析[J]. 应用与环境生物学报, 2014, 20(5): 832-839. GUO Ya-ping, ZHANG Guo-qing, CHEN Qing-jun, et al. Bacterial community structure analysis for mushroom(Agaricus bisporus)compost using PCR-DGGE technique[J]. Chinese Journal of Applied and Environmental Biology, 2014, 20(5): 832-839. |
| [22] |
许修宏, 成利军, 许本姝, 等. 基于高通量测序分析牛粪堆肥中细菌群落动态变化[J]. 东北农业大学学报, 2018, 49(3): 10-20. XU Xiu-hong, CHENG Li-jun, XU Ben-shu, et al. Analysis of bacterial community dynamics in cow manure composting using high throughput sequencing[J]. Journal of Northeast Agricultural University, 2018, 49(3): 10-20. |
| [23] |
Pankratov T A, Ivanova A O, Dedysh S N, et al. Bacterial populations and environmental factors controlling cellulose degradation in an acidic Sphagnum peat[J]. Environmental Microbiology, 2011, 13(7): 1800-1814. DOI:10.1111/j.1462-2920.2011.02491.x |
| [24] |
Ma S, Fang C, Sun X X, et al. Bacterial community succession during pig manure and wheat straw aerobic composting covered with a semipermeable membrane under slight positive pressure[J]. Bioresource Technology, 2018, 259: 221-227. DOI:10.1016/j.biortech.2018.03.054 |
| [25] |
郁聪.太平洋海洋杆菌(Oceanobacillus pacificus sp. nov.)等5株南太平洋新菌的分类鉴定及相关特性分析[D].青岛: 中国海洋大学, 2014. YU Cong. Taxonomic analysis and related research of Oceanobacillus pacificus sp. nov. and other five novel bacteria from the South Pacific Gyre[D]. Qingdao: Ocean University of China, 2014. |