 2020, Vol. 39
2020, Vol. 39



2. 中国农业科学院农业环境与可持续发展研究所农业农村部农业环境重点实验室, 北京 100081
2. Institute of Environment and Sustainable Development in Agriculture, Chinese Academy of Agricultural Sciences/Key Laboratory of Agro-Environment, Ministry of Agriculture and Rural Affairs, Beijing 100081, China
水铁矿因其较小的颗粒尺寸、巨大的比表面积和无定形的多孔隙结构而对砷表现出极强的吸附作用,通常用于土壤砷污染的原位修复[1-4]。但水铁矿本身的弱结晶态结构极易受到土壤环境因素(土壤pH、Eh、溶质离子组成、土壤温度及部分土壤理化性质等)的影响而向更加稳定的二级产物(如针铁矿和赤铁矿)发生转化[5]。这些因素都会一定程度上影响水铁矿在土壤中的稳定性,进而导致水铁矿结合的砷再次释放到土壤环境中造成二次污染。
在土壤环境中,土壤水分的差异能够显著影响水铁矿的解离/转化过程。已有部分研究结果表明了水铁矿在土壤高水分环境下结构不稳定[5-7],随着土壤水分的增加,土壤逐渐进入还原态,还原状态下Fe(Ⅱ)的存在可以强烈地促进水铁矿的解离作用从而破坏水铁矿的结构,导致其表面吸附的As(Ⅴ)再次释放到土壤中[7-9]。砷的形态对土壤氧化-还原环境的变化十分敏感,土壤的干湿交替循环过程对于砷在土壤孔隙水的迁移动力学过程有着显著的影响[10-12]。在土壤还原状态下释放至土壤溶液的As(Ⅴ)被还原成毒性更高的As(Ⅲ),提高了砷在环境中的风险危害。陈丽娜[13]通过对水稻田不同水分管理模式下的研究发现,铁-砷之间的变化关系随着干湿交替的水分变化呈现出周期性变化,而这种周期性变化主要由于土壤水分的变化引起土壤Eh和pH的变化,进而影响水稻根际铁膜对砷的阻隔作用及稻田土壤中砷的生物有效性。部分学者研究发现砷在土壤还原条件下的移动性明显增加[10-11, 14-15]。但合理控制土壤水分不达到长期淹水条件,可以提高土壤中铁氧化物的稳定性,能够显著降低土壤中砷的移动性和生物有效性[11, 15-16]。
连续提取法目前广泛用于分析土壤中重金属的形态。Wenzel等[17]提出的五步连续提取法已经证明了其能够有效分离砷在土壤中的五种不同形态(弱酸结合态、碳酸盐结合态、弱结晶态铁锰氧化物结合态、稳定结晶态铁锰氧化物结合态和残渣态)。部分学者通过对该方法进一步探究,发现通过分析第三和第四步两种形态可以准确表征水铁矿结构变化过程中砷的结合形态的迁移过程,为进一步研究水铁矿稳定性变化中砷-铁的结合规律提供一定的方法支持[7, 18]。此外,薄膜梯度扩散技术(DGT)近年来因其对重金属在土壤环境中便捷准确的表征模式也广泛应用于重金属元素在土壤中的原位迁移转化过程,其结果能从另一层面分析砷-铁在土壤中原位同步释放的过程。受限于传统的化学提取法,目前关于水铁矿在土壤中稳定性的变化过程对砷影响机制的研究仍然较少,另外土壤水分条件的差异对水铁矿钝化效果的影响过程也鲜有报道。因此,本文通过结合连续提取法和薄膜梯度扩散技术(DGT)来探究不同水分管理模式下水铁矿在土壤中稳定性的变化过程中对砷-铁结合方式的影响,并从动力学角度分析外源水铁矿添加后砷在土壤中的迁移规律,为在不同水分管理模式下合理利用水铁矿作为砷污染土壤钝化剂提供一定的技术方法和理论依据。
1 材料与方法 1.1 供试土壤本研究中三种土壤均采自湖南石门县,三种土壤为典型红壤母质发育下形成的第四纪红壤、石灰岩红壤和板页岩红壤。根据《土壤环境质量农用地土壤污染风险管控标准(试行)》(GB 15618—2018)将三种采集于石门雄黄矿区附近的三种不同砷浓度的农田表层土壤分为高浓度风险(High risk,HR)、中浓度风险(Medium risk,MR)和无风险土壤(No risk,NR)。土壤的基本理化性质见表 1。采集的土壤经过自然风干后均匀混合,去除土壤中较大的植物残体和沙砾、石块等杂物,过2 mm尼龙网筛后备用。
|
|
表 1 供试土壤的理化性质 Table 1 Physical and chemical properties of soils |

本研究所用水铁矿采用人工合成的方式获得[19],具体合成方法如下:FeCl3·6H2O溶解于超纯水中,随后向FeCl3溶液中加入1 mol·L-1 KOH溶液并且调节pH至7~8,合成过程中使用磁力搅拌器确保FeCl3和KOH溶液充分混合均匀,此反应进行大约30~40 min后即可获得红褐色水铁矿胶体悬浊液。随后将水铁矿悬浊液分装至若干50 mL离心管,3500 r·min-1转速下离心10 min,随后倒掉上清液,加入超纯水让离心管底部的水铁矿胶体再次悬浊后用往复振荡器以120 r·min-1振荡30 min,离心10 min。如此反复三次纯化水铁矿胶体。洗涤三次后将水铁矿胶体导入透析袋中透析7~10 d直至电导率降至10 μS·cm-1。随后冷冻干燥透析后的水铁矿胶体,得到黑褐色粉末,保存于干燥器中待用。
1.3 试验设计三种供试土壤中外源添加水铁矿的量均为0.1%(m/m),并同时设置三组重复。土壤在三种不同水分条件下培养:持续30%最大田间持水量(Soil water holding capacity,SWHC)、持续100%SWHC和干湿交替循环(Dry / wet cycle, DWC)。干湿交替循环在30%SWHC和100%SWHC之间进行干湿交替循环试验,具体处理过程如下:每组循环周期为11 d(2 d控制为100%SWHC,7 d控制为30%SWHC,2 d为在室温条件下从100%SWHC到30%SWHC自然晾干的时间)。第一循环首先在100%SWHC条件下培养2 d,然后晾干至30%SWHC,保持30%SWHC培养7 d(第11 d),完成第一循环的干湿交替。然后根据称重法加水至100%SWHC依次进行第二轮和第三轮干湿交替土壤培养过程。整个过程共进行三轮干湿交替循环。在2、11、13、22、24、33 d取部分土壤用MBL-DGT(Chelex-TiO2混合吸附相DGT,用于同时分析测定砷-铁)测定土壤中活性态砷和铁浓度的变化。同时在第11、22、33 d取部分土壤样品分析土壤溶液中化学提取态的砷浓度变化。在第2 d和第33 d分析外源添加水铁矿在土壤中的转化及水铁矿添加后土壤中砷赋存形态的变化过程。土壤在持续30%SWHC和100%SWHC水分条件下培养时间周期及取样时间与DWC相同。
1.4 测定项目及方法 1.4.1 活性态砷、铁的测定及计算DGT在土壤中的放置测定过程和计算方法参照Wang等[20]。称取培养过程中土壤样品60 g,随后加水至80%~100%田间最大持水量。均匀搅拌使土壤成泥浆状,随后放置1 d使土水平衡。在放置DGT前,将平衡后土壤均匀分成三份,放入三个不同的小塑料培养皿中,然后进行DGT放置试验。DGT放置试验开始前12 h将DGT装置从4 ℃冰箱中取出,取出后用洗瓶反复冲洗DGT窗口。先在DGT装置表面暴露的窗口(滤膜)上均匀地涂抹一些糊状的土壤,用手将DGT轻轻旋转挤压进土壤(约1 cm深),确保DGT装置与土壤之间接触良好。立即记录DGT施用于土壤中的时间以及施用过程中环境的温度,尽量满足温度变化在±2 ℃以内。
DGT中砷和铁的洗脱步骤和计算方法参照Zhang等[7]。从土壤中取出DGT后,用超纯水清洗其窗口表面沾染的土壤,随后打开DGT装置,小心取出装置最底层的吸附凝胶层,先用1 mL 1 mol·L-1硝酸溶液洗脱24 h,用于洗脱Chelex吸附的铁。随后取出凝胶用超纯水反复冲洗,用1 mL 1 mol·L-1氢氧化钠溶液洗脱24 h,用于洗脱TiO2吸附的砷。待两组洗脱液均平衡后,将两组洗脱液混合、稀释后,用电感耦合等离子体质谱仪(Inductively coupled plasma - mass spectrometry,ICP-MS)分析测定洗脱液中砷和铁的浓度。
元素在DGT吸附相上富集的质量(m,µg)和活性态浓度(CDGT,µg·L-1)按照公式(1)和(2)进行计算:
| $ $ | (1) |
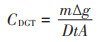
|
(2) |
式中:Ce表示洗脱液中重金属的浓度,μg·L-1;Vgel表示吸附相胶的体积,mL;Vacid表示加入酸的体积,通常1 mL;fe为洗脱效率;∆g为扩散相的厚度,cm;D为目标元素的扩散系数,cm2·s-1;t为DGT的放置时间,s;A为DGT的窗口面积,cm2。
1.4.2 水铁矿的形态转化以及其转化过程中结合态砷的测定水铁矿、结晶态铁氧化物及不同形态砷的含量测定采用两步连续提取法进行操作[18]。方法步骤如表 2所示。
|
|
表 2 土壤中不同铁氧化物结合态砷和不同结晶态铁氧化物的分级提取步骤 Table 2 Two-step sequential extraction of varied fractionation As and crystalline iron oxide |

为了解不同水分条件下水铁矿添加前后以及随时间变化过程中土壤砷的迁移动力学特征,本试验结合DGT测定土壤活性态砷(CDGT)、土壤液相砷(土壤溶液砷,Csoln)和土壤固相结合态砷(NaHCO3提取态砷,Cs)的相关测定值计算土壤R值(固相向液相的再补给能力,R=CDGT/Csoln,0 < R < 1)和砷在土壤固液两相间的分配系数Kd(Kd=Cs/Csoln,cm3·g-1),具体计算结果见表 6。NaHCO3对弱结合态砷的提取方法参照Wang等[21]。称取1 g土,加入25 mL 0.5 mol·L-1 NaHCO3后振荡2 h,离心,上层清液过滤后待测。土壤溶液中提取砷参照Wang等[21]。将DGT测定后的土壤装入离心管中,在离心机5000 r·min-1条件下离心10 min,随后分离上清液,过滤后待测。利用电感耦合质谱仪(ICP-MS)对化学提取态砷和土壤溶液中的砷进行测定。
|
|
表 6 不同水分管理模式下土壤中添加水铁矿11、22、33 d后化学提取态砷的变化(mg·kg-1) Table 6 Variation of chemical extractable As at varied water management modes after ferrihydrite addition 11, 22 d and 33 d(mg·kg-1) |
土壤溶液中Fe(Ⅱ)采用邻菲罗啉比色法测定。培养条件下取土20 g(湿质量)后离心,分离上层清液并过0.45 μm孔径滤膜后待测。分别吸取0、0.1、0.2、0.4、0.6、0.8、1 mL Fe(Ⅱ)标准贮备液(100 μg·L-1)于10 mL容量瓶中,随后加入0.2 mL盐酸羟胺溶液、1 mL醋酸-醋酸铵溶液以及0.1 mL邻菲罗啉溶液并加水定容至刻度线,此时得到浓度梯度为0、1、2、4、6、8、10 μg·L-1。放置15 min显色,用1 cm比色皿,以空白吸光度为参比,测定各溶液的吸光度。土壤溶液的测定参照标准溶液的测定方法。绘制标准溶液吸光度曲线及方程(吸光度A-二价铁离子浓度CFe(Ⅱ)),通过该方程计算不同样品中Fe(Ⅱ)的浓度。
1.5 数据处理试验中所得数据均采用Excel 2007进行处理分析,同时利用SPSS 21.0进行相关数据的统计分析。
测定土壤中砷和铁在DGT扩散凝胶层中的扩散系数来自Zhang等[22]和Luo等[23]测定或提及的试验数据,用温度记录仪记录试验过程中的温度变化,采用试验过程中平均温度下的扩散系数进行计算。
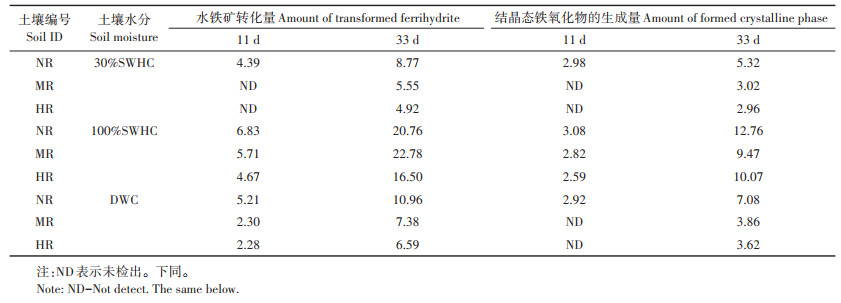
2 结果与分析 2.1 外源水铁矿的转化及不同铁氧化物结合态砷的变化水铁矿向结晶态铁氧化物的转化量如表 3所示。从表 3可知,水铁矿在三种土壤100%SWHC和DWC条件下培养均在11 d内发生一定程度的转化,但在30%SWHC条件下,仅在土壤NR中发生转化,而在土壤MR和HR中水铁矿在11 d内均未发生转化。在100%SWHC的高水分条件下培养33 d时,分别有22.78%、20.76%和16.50%水铁矿在土壤MR、NR和HR发生转化,显著高于其在同时期DWC和30%SWHC条件下的转化量。伴随着水铁矿的转化,三种水分条件下均有结晶态铁氧化物生成,但相比于100%SWHC,30%SWHC和DWC条件下结晶态铁氧化物在三种土壤中的生成量较低,在11 d时几乎无法测出,仅在33 d时有少量的结晶态铁氧化物可以测定。
|
|
表 3 不同水分管理模式下水铁矿添加11、33 d后弱结晶态铁氧化物的转化量和结晶态铁氧化物的生成量(%) Table 3 Transformation of poor crystalline iron oxide phase to well crystalline iron oxide phase after ferrihyrite addition 11 d and 33 d at varied water management modes(%) |
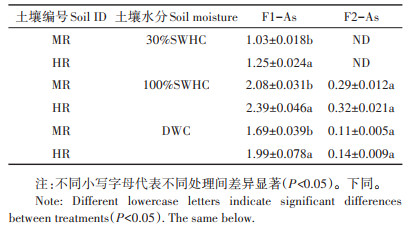
水铁矿添加33 d后,弱结晶态铁氧化物结合态砷(F1-As)和结晶态铁氧化物结合态砷(F2-As)在两种砷污染土壤中均呈现出增加的趋势(表 4)。其中在100%SWHC条件下时F1-As形态中的砷含量要高于DWC和30%SWHC条件下。水铁矿添加33 d后,在30%SWHC条件下培养F2-As形态中的砷含量并未检出,但两种土壤在100%SWHC和DWC条件培养下,F2 -As结合态砷的含量均少量增加,其中在100%SWHC条件下时F2-As形态中砷的增加量大于DWC水分条件下的增加量。但对比两种土壤MR和HR可知,在三种水分条件下,土壤HR中F1-As形态的砷显著高于土壤MR(P < 0.05)。另外在两种不同水分条件下,两种土壤间F2-As形态砷的量并无显著性差异(P>0.05)。
|
|
表 4 不同水分管理模式下水铁矿添加33 d后弱结晶态铁氧化物结合态砷(F1-As)和结晶态铁氧化物结合态砷(F2-As)的增加量(mg·kg-1) Table 4 Increment of poor crystalline iron oxide phase combined As(F1-As)and well crystalline iron oxide phase combined As (F2-As)after ferrihydrite addition 33 d at varied water management modes(mg·kg-1) |
外源水铁矿添加后,土壤中活性态铁的浓度变化过程如图 1所示。相比于未添加水铁矿的处理,活性态铁的浓度在三种土壤不同水分条件下均在开始阶段表现出增加趋势,但其变化趋势之间有明显差异。如在土壤NR中,水铁矿在100%SWHC条件培养过程中活性态铁在33 d从初始的102.23 μg·L-1持续升高至242.12 μg·L-1。但在30%SWHC条件下活性态铁的浓度仅在13 d左右出现峰值(158.48 μg·L-1)后便呈现出明显的下降趋势。在DWC条件下土壤MR中活性态铁的浓度变化过程随水分变化表现出一定交替周期性变化。在30%SWHC条件下培养,在水铁矿加入初期,活性态铁浓度变化与100%SWHC和DWC条件一样均表现出较为快速的上升趋势,随后在25 d左右与30%SWHC一样活性态铁表现出明显的持续下降趋势直至33 d结束。三种水分条件下活性态铁的浓度高低顺序为100%SWHC>DWC>30%SWHC。在土壤MR和土壤HR的三种水分处理中,30%SWHC和DWC条件下也表现出与土壤NR相似的变化趋势。但二者与土壤NR不同的是在100%SWHC条件下培养进入第三周期开始时活性态铁的浓度便开始表现出下降的趋势而非持续上升趋势。

|
图 1 不同水分管理模式下活性态铁随老化时间的变化 Figure 1 Variation of labile Fe at varied water management modes after ferrihydrite addition |
外源水铁矿添加后土壤中活性态砷的变化如图 2所示。水铁矿添加后两种土壤中活性态砷的浓度都显著降低。相比于未添加水铁矿处理,水铁矿添加第2 d时活性态砷浓度在土壤MR和土壤HR三种不同水分条件下降低程度分别到达26.91%~42.08%和39.33%~60.75%,其中在100%SWHC条件下活性态砷浓度下降程度最大,但这种下降的趋势在100%SWHC和DWC条件下仅维持到22 d。培养22 d后,活性态砷的浓度开始表现出缓慢的上升趋势,表明砷从水铁矿表面解吸并释放至土壤环境中。

|
图 2 不同水分管理模式下活性态砷随老化时间的变化 Figure 2 Variation of labile As at varied water management modes after ferrihydrite addition |
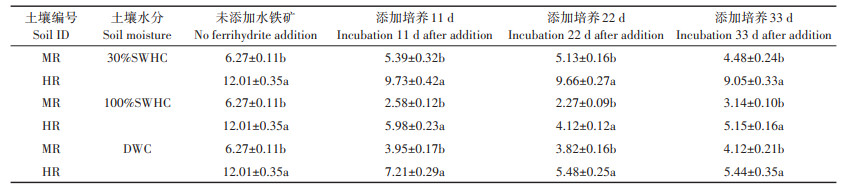
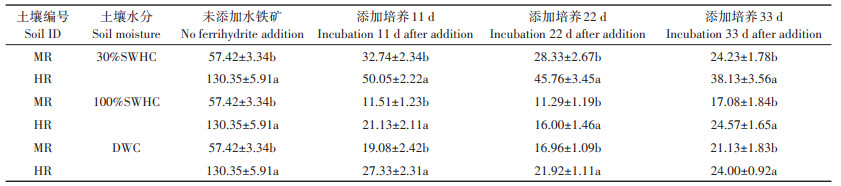
土壤溶液砷的变化过程如表 5所示。水铁矿加入后,在三种不同水分条件下土壤溶液砷的浓度均显著降低,但在三种水分条件下表现出的趋势却有所差异,其中在100%SWHC和DWC条件下降幅要明显高于30%SWHC。相比于培养第0 d未添加水铁矿时,在30%SWHC条件下,水铁矿加入后土壤溶液中砷浓度在33 d内表现为持续下降的趋势。在100%SWHC和DWC条件下,土壤溶液中砷在开始的22 d内浓度持续降低,但在33 d时浓度相比于22 d时略有升高。化学提取态砷在三种水分条件下变化过程与土壤溶液砷变化过程相似(表 6)。相比于培养第0 d未添加水铁矿时,在30%SWHC条件下,水铁矿加入后化学提取态砷浓度在33 d内仍表现为持续下降趋势,而在100%SWHC和DWC条件下,水铁矿加入后的22 d化学提取态砷浓度均明显降低,但在33 d时,相比于22 d其浓度又表现出微弱的升高的趋势。这两部分的结果与活性态砷的浓度变化规律基本一致。
|
|
表 5 不同水分管理模式下土壤中添加水铁矿11、22、33 d后土壤溶液中砷的变化(mg·kg-1) Table 5 Variation of water extractable As at varied water management modes after ferrihydrite addition 11, 22 d and 33 d(mg·kg-1) |
土壤Eh值和Fe(Ⅱ)浓度随时间变化曲线如图 3所示。土壤三种不同水分条件下培养过程中Eh表现出三种截然不同的变化趋势。在30%SWHC条件下,三种土壤Eh值在33 d内平均维持在450~480 mV左右小幅波动。在100%SWHC条件下,三种土壤Eh值在0~33 d内表现出明显的下降趋势,均从450 mV左右降至160~180 mV。在DWC条件下,三种土壤的Eh值则随着干湿交替的变化也呈现出明显的周期性下降-上升-下降-上升的趋势,这种交替变化过程一直保持到第三循环周期结束。

|
图 3 土壤Eh和土壤溶液中Fe(Ⅱ)浓度在33 d随时间的变化 Figure 3 Variation of Fe(Ⅱ)concentration in soil solution and Eh in three soils at varied SWHCs after ferrihydrite addition |
土壤溶液中Fe(Ⅱ)浓度的变化过程如图 3所示。从图中可知,在三种土壤中,仅有在100%SWHC条件下Fe(Ⅱ)浓度表现出明显的上升趋势,而在30%SWHC和DWC条件下,Fe(Ⅱ)浓度几乎无法测出。如在土壤NR 100%SWHC条件下,Fe(Ⅱ)浓度在33 d内迅速从0上升至4.65 mg·L-1,而在30%SWHC和DWC条件下则无法测出Fe(Ⅱ)浓度变化。在两种砷污染土壤中也表现出类似的趋势。在土壤MR和土壤HR中,Fe(Ⅱ)浓度在33 d内也分别从0上升至6.56 mg·L-1和4.89 mg·L-1,而在30%SWHC和DWC水分条件下,Fe(Ⅱ)浓度也仅维持较低浓度,无明显上升趋势。
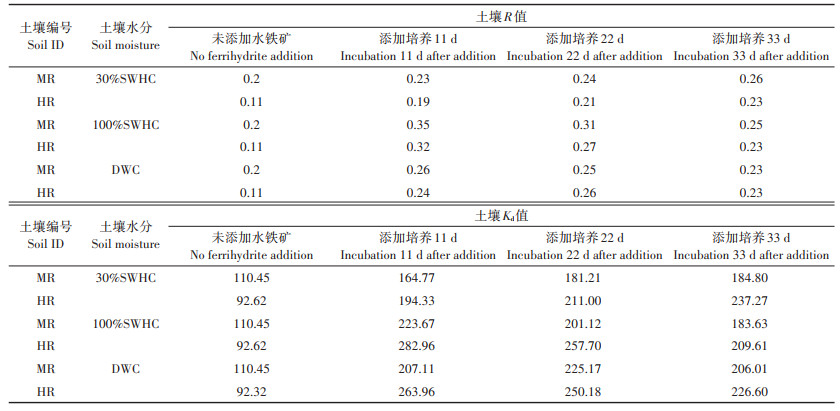
2.5 水铁矿添加后对砷迁移动力学的影响从表 7可知,两种砷污染土壤中,外源水铁矿添加后土壤的R值比未添加水铁矿时均明显升高,但随着水铁矿在土壤中的老化,三种不同水分条件下R值也表现出不同的变化趋势。在30%SWHC条件下,随着水铁矿老化时间的进行,R值从加入的第11 d开始直至33 d结束,保持持续上升的趋势,但在100%SWHC和DWC条件下,两种土壤R值在水铁矿添加的11 d后显著升高,但随后都开始呈现下降的趋势。两种土壤在不同水分条件下Kd值在一定程度表现出与R值变化相对应的趋势。当水铁矿添加后,所有处理中砷的Kd值均显著升高,但在不同水分条件下,两种土壤Kd值的变化仍有差异。在30%SWHC条件下,Kd值在33 d内呈逐渐升高的趋势。而在100%SWHC和DWC条件下,在初始阶段Kd值明显升高,但随后与R值变化类似,表现出逐渐下降的趋势。
|
|
表 7 不同水分管理模式下外源水铁矿添加后砷在土壤中的迁移动力学参数 Table 7 Kinetic parameters of As under varied water management modes in soils before/after ferrihydrite |
三种水分管理模式下,水铁矿不论转化速率还是解离速率(活性态铁的变化过程)的大小顺序均为100%SWHC>DWC>30%SWHC。土壤水分的高低主要调控了土壤Eh和pH值的变化。此外在高水分条件下,土壤溶液的增加会一定程度上促进水溶性离子的解离过程。因此在100%SWHC条件下,一些离子如H+和Fe(Ⅱ)可能会在加速水铁矿转化/解离过程中起主要作用[6-7]。本研究的三种供试土壤均为酸性(pH 4~4.96),因此H+在一定程度对土壤中水铁矿转化的过程有比较强烈的影响。前人已有研究表明,水铁矿在酸性条件下,H+能够明显加速其转化[5, 7, 24]。Schwertmann等[25]认为H+会通过形成一系列的中间产物促进水铁矿的解离。FeOOH在酸性条件下倾向于受到H+的质子化作用并且形成中间产物如Fe(OH)2+ [pH>4,FeOOH+H+→Fe(OH)2+]或者Fe(OH)2+[pH < 4,FeOOH+2H+→Fe(OH)2++H2O]。这样的Fe(Ⅲ)中间产物会通过晶型结构重组的作用逐渐形成结晶态铁氧化物。
除H+的质子化作用外,在100%和30%SWHC条件下水铁矿解离/转化之间的差异表明,当土壤逐渐进入到部分还原状态时,产生的Fe(Ⅱ)也可能很大程度上催化了水铁矿的转化。大量研究表明即使在室温条件下Fe(Ⅱ)也能够显著提高水铁矿向结晶态铁氧化物的转化速率[26-29]。土壤中Fe(Ⅱ)的来源为生物或非生物过程[30],并且在酸性土壤环境中Fe(Ⅱ)的稳定性和溶解性会显著增加,甚至在有少量氧气存在的情况下也可大量存在[31]。这表明在本研究中,酸性还原状态的土壤中,溶解态Fe(Ⅲ)很大程度上在土壤微孔中被还原成Fe(Ⅱ)[32-33]。在土壤高水分条件下Fe(Ⅱ)会成为加速水铁矿转化的主要动力[18, 34],生成的Fe(Ⅱ)吸附在水铁矿表面并且作为还原剂进行电子传递作用[27]。水铁矿的结构会因为一个电子的注射作用导致破坏发生重组,电子进入内部结构后使水铁矿变得更加不稳定并且催化水铁矿释放出Fe(Ⅱ),由于这种反应过程是一个Fe(Ⅱ)单位吸附在水铁矿表面,并引起一个Fe(Ⅱ)单位释放,因此可以理解为在整个催化反应过程中Fe(Ⅱ)的总量维持恒定[28]。因此水铁矿在100%和30%SWHC培养条件下所表现出的差异可能主要来自于Fe(Ⅱ)的催化作用。图 3中的结果也表明了Fe(Ⅱ)浓度在三种土壤100%SWHC条件下随着Eh值降低明显升高,而30%SWHC和DWC条件Fe(Ⅱ)几乎无法检测。在本研究中,上述原因也合理地解释了在三种土壤中30%SWHC条件下活性态铁在短时间内到达峰值后便表现出下降的趋势,而由于Fe(Ⅱ)在持续100%SWHC条件下的土壤中不断地催化水铁矿的解离导致活性态铁的浓度持续升高,直至24 d左右才出现下降的趋势。综上所述,可以发现水铁矿在30%SWHC条件下转化速率的快慢更多受到土壤初始pH的影响,而在100%SWHC条件下,在水铁矿加入初期受到pH(H+)作用,而随着土壤还原态的逐渐形成,Fe(Ⅱ)成为控制水铁矿转化速率快慢的主导因素。此外,本研究中发现水铁矿在DWC条件下的变化过程更接近于30%SWHC培养条件,这可能是由于在DWC水分试验设置中模拟了旱地灌溉后的田间水分变化(灌溉后土壤在夏季处于100%SWHC的时间极少,仅为2~3 d左右),而土壤处于30%SWHC条件下的时间较长。因而在这样干湿交替过程中,土壤几乎无法进入还原状态,几乎不会有Fe(Ⅱ)催化水铁矿转化。但水铁矿在其中的转化/解离速率仍然高于30%SWHC,并且活性态铁随水分呈显著周期性变化,这可能是由于短暂的高水分条件下H+的质子化作用要强于低水分条件下的作用。
3.2 不同水分管理模式下水铁矿稳定性变化对砷在土壤中迁移性的影响在水铁矿加入的初期,活性态砷、土壤溶液中砷、化学提取态砷的浓度在三种水分条件下均显著降低,并且在DWC和100%SWHC条件下培养对砷表现出同样的吸附能力。其主要原因可能是在土壤水分较为充足的条件下增加了水铁矿表面吸附位点与砷的接触,提高了水铁矿对砷吸附效率[7, 35]。有研究报道水铁矿对砷的吸附过程十分迅速,在纯净溶液体系下短短数小时内就可达到极高的吸附量[36],即使在土壤环境中其对砷的吸附也在短短几天也会趋于平衡[37-38]。因此,水铁矿在DWC和100%SWHC条件下培养开始的2 d内土壤水分含量均为100%SWHC时,水铁矿在土壤溶液中对大部分活性态的砷吸附过程已经接近完成。砷被水铁矿吸附后,与水铁矿以共沉淀的方式或者随水铁矿的转化被稳定结合到稳定结晶态铁氧化物中(如纤铁矿和针铁矿)[39-40]。除水铁矿的吸附作用外,本研究表明伴随着水铁矿向稳定结晶态铁氧化物的转化,也有部分F1-As向F2-As迁移进而形成更稳定形态的砷(表 4),这也表明砷可能随水铁矿的转化被结合得更加稳定。由于F2-As形态含量有所增加,可以认为砷可能一方面与新生成的结晶态铁氧化物通过表面吸附作用的方式结合,也有可能进入新生成的铁氧化物内部参与结构的组成,其中何种作用方式占主导仍需进一步研究。
尽管水铁矿在土壤中表现出对砷较高的吸附效果,但在本研究中仍然发现随着水铁矿的转化/解离形成新的稳定结晶态的铁氧化物会影响其对砷的吸附效率,导致砷向土壤环境的二次释放。Ford[40]指出水铁矿的转化对砷在土壤中的化学行为可能有以下几方面影响:(1)水铁矿转化后砷仍然吸附在水铁矿表面或者吸附在新生成的结晶态铁氧化物表面;(2)由于其比表面积逐渐减少或者吸附位点逐渐缺失,砷可能从水铁矿表面解吸;(3)砷可能随着新的结晶态铁氧化物的生成而参与新的铁氧化物的结构组成。有研究报道在土壤好氧状态下As(Ⅴ)与水铁矿以内表面的方式结合[41]。在本研究中,在30%SWHC条件下土壤处于高度好氧状态,土壤中的砷以As(Ⅴ)为主要形态,在这种情况下水铁矿的解离速率一方面较慢,而另一方面As(Ⅴ)与水铁矿的结合方式又相对稳定,因此并未观察到土壤中活性态砷、化学提取态砷和土壤溶液中的砷含量升高的趋势。但在Zhang等[7]的研究报道中30%SWHC条件下活性态砷的浓度也有所增高,原因可能是土壤处于高度好氧状态下磷(P)和As(Ⅴ)的竞争,本研究中并未出现这样的变化趋势,可能是因为本研究中两种砷污染土壤的有效磷含量(14.32 mg·kg-1和18.56 mg·kg-1)明显低于Zhang等[7]实验中土壤有效磷含量(78.32~86.56 mg·kg-1)。
在100%SWHC和DWC条件下,活性态砷、化学提取态砷和土壤溶液中的砷在土壤MR和HR水铁矿添加20 d后均表现出持续升高的趋势。尽管DWC条件下水铁矿的解离转化过程并不明显,但三种形态砷浓度升高的原因很有可能是干湿交替循环周期过程中短暂地维持100%SWHC条件时,H+对水铁矿的解离作用要明显强于持续30%SWHC培养条件,因而导致在DWC条件下仍有少量砷的释放。而土壤处于持续100%SWHC高水分条件下三种形态砷浓度的增加量高于DWC,可理解为除H+质子化作用外,还与水铁矿在部分还原状态(Eh为250 mV左右)下的解离作用有关,该结果与Al-Abed等[42]研究结果基本一致,其研究结果表明当Eh值逐渐从470 mV降低至250 mV时,溶解态砷和溶解态铁的浓度表现出明显的快速上升趋势。Nielsen等[38]报道砷在土壤中释放过程与季节性的水分变化有很大关系。在高度氧化态土壤中砷的释放较低,而在高水分条件下,水铁矿的解离过程发生十分迅速,同时伴随砷快速释放至土壤溶液中。在土壤处于好氧状态时,仅H+对水铁矿的解离作用,水铁矿表面仍然可以吸附足够多的砷,但土壤进入还原状态时Fe(Ⅱ)也参与到对水铁矿的解离,导致其表面无法吸附更多的砷,因而引起了活性态砷浓度的升高。
3.3 土壤砷迁移动力学参数的应用及合理水分调控意义添加水铁矿后,土壤固相中砷向液相的再补给能力(R值)无论在何种水分条件下均明显升高,但随后在高水分条件下又均呈现出下降的趋势。土壤固液分配系数Kd也表现出类似相关的变化过程。由于水铁矿的加入,极大地降低了土壤液相砷,从而导致土壤固相中的砷(Cs)相比液相中的砷(Csoln)比例有所增加,导致Kd值在水铁矿添加后激增,但随着部分水铁矿的转化/解离以及新的结晶态铁氧化物生成,土壤液相中砷的浓度再次升高,Kd值短暂下降。通过对动力学参数的把控,进一步合理调控水分是控制水铁矿向稳定结晶态铁氧化物转化的关键。在本研究中,尽管水铁矿作为一种潜在的高效吸附材料能够显著降低土壤中活性态的砷,并且显著改变了R值和Kd值,但在高水分条件下(如100%SWHC)其结构的不稳定性导致砷向土壤环境中二次释放。已有的研究结果显示水铁矿在砷污染土壤中如处于低水分条件下其转化速率极慢,需要10年之久,而在中度湿润的水分条件下(70%SWHC)完全转化只需要2~4年[7]。因此在修复旱地农田时,耕作条件允许的情况下,合理地调控土壤处于低水分状态(如30%~50%SWHC)将有助于保持水铁矿的稳定性,使其长期处于对砷高效稳定吸附状态。旱地土壤在灌溉期经历的干湿交替过程也一定程度上存在砷释放的风险,尽管水铁矿在其中的稳定性变化过程更接近于30%SWHC(相对转化解离速率比较慢),但在酸性土壤中H+的质子化作用导致的短暂高水分条件下砷向土壤环境中的释放仍然需要引起砷污染农田修复的重视。
4 结论(1)水铁矿在土壤中添加33 d后均发生了转化/解离的过程,并且其转化/解离速率显著受到了水分的调控,在高水分条件(100%SWHC)下由于H+和Fe(Ⅱ)的同时作用,其结构稳定性受到了威胁。尽管外源水铁矿添加后在所有水分管理模式中均显著降低活性态砷、化学提取态砷和土壤溶液中砷的浓度,但在100%SWHC和DWC条件下因水铁矿的转化/解离过程的发生,导致在两种砷污染土壤中三种形态的砷浓度均出现升高的趋势。表明水铁矿的转化/解离在一定程度上导致了砷向土壤溶液二次迁移释放。
(2)尽管水铁矿的不稳定性引起了砷向土壤环境中的释放,但在100%SWHC和DWC条件下在两种砷污染土壤中弱结晶态铁氧化物结合态砷(F1-As)的含量均显著增加,并且随着水铁矿向结晶态铁氧化物的转化而逐渐向更稳定的形态(F2-As)迁移,表明部分砷最终会伴随着水铁矿向稳定的结构转化而结合得更加紧密。
(3)迁移动力学结果也进一步表明合理调控土壤水分是确保水铁矿长效吸附砷的关键。在保证农业生产要求的基础上,中高浓度的砷污染农田修复并进行复耕后,应尽量采用耕地旱作方式,维持土壤中低水分条件(如30%~50%SWHC),可以一定程度上保证水铁矿的钝化效果。
| [1] |
林志灵, 曾希柏, 张杨珠, 等. 人工合成铁, 铝矿物和镁铝双金属氧化物对土壤砷的钝化效应[J]. 环境科学学报, 2013, 33(7): 1953-1959. LIN Zhi-ling, ZENG Xi-bai, ZHANG Yang-zhu, et al. Immobilization effects of synthetic Fe/Al minerals and Mg/Al layered double oxides (Mg/Al-LDO)additions on arsenic(As)in soils[J]. Acta Scientiae Circumstantiae, 2013, 33(7): 1953-1959. |
| [2] |
钟松雄, 尹光彩, 何宏飞, 等. 不同铁矿物对水稻土砷的稳定化效果及机制[J]. 环境科学学报, 2017, 37(5): 1931-1938. ZHONG Song-xiong, YIN Guang-cai, HE Hong-fei, et al. Stabilization effect of arsenic by different iron minerals in paddy soils and the related mechanism[J]. Acta Scientiae Circumstantiae, 2017, 37(5): 1931-1938. |
| [3] |
Abad-Valle P, Álvarez-Ayuso E, Murciego A. Evaluation of ferrihydrite as amendment to restore an arsenic-polluted mine soil[J]. Environmental Science and Pollution Research, 2015, 22(9): 6778-6788. DOI:10.1007/s11356-014-3868-6 |
| [4] |
Sun Y, Liu R, Zeng X, et al. Reduction of arsenic bioavailability by amending seven inorganic materials in arsenic contaminated soil[J]. Journal of Integrative Agriculture, 2015, 14(7): 1414-1422. DOI:10.1016/S2095-3119(14)60894-7 |
| [5] |
Zhang T, Zeng X B, Zhang H, et al. Investigation of synthetic ferrihydrite transformation in soils using two-step sequential extraction and the diffusive gradients in thin films(DGT) technique[J]. Geoderma, 2018, 321: 90-99. DOI:10.1016/j.geoderma.2018.01.034 |
| [6] |
Yamaguchi N, Nakamura T, Dong D, et al. Arsenic release from flooded paddy soils is influenced by speciation, Eh, pH, and iron dissolution[J]. Chemosphere, 2011, 83(7): 925-932. DOI:10.1016/j.chemosphere.2011.02.044 |
| [7] |
Zhang T, Zeng X B, Zhang H, et al. The effect of the ferrihydrite dissolution/transformation process on mobility of arsenic in soils:Investigated by coupling a two-step sequential extraction with the diffusive gradient in the thin films (DGT)technique[J]. Geoderma, 2019, 352: 22-32. DOI:10.1016/j.geoderma.2019.05.042 |
| [8] |
Nickson R T, McArthur J M, Ravenscroft P, et al. Mechanism of arsenic release to groundwater, Bangladesh and West Bengal[J]. Applied Geochemistry, 2000, 15(4): 403-413. DOI:10.1016/S0883-2927(99)00086-4 |
| [9] |
Hansel C M, Benner S G, Neiss J, et al. Secondary mineralization pathways induced by dissimilatory iron reduction of ferrihydrite under advective flow[J]. Geochimica et Cosmochimica Acta, 2003, 67(16): 2977-2992. DOI:10.1016/S0016-7037(03)00276-X |
| [10] |
Chatain V, Sanchez F, Bayard R, et al. Effect of experimentally induced reducing conditions on the mobility of arsenic from a mining soil[J]. Journal of Hazardous Materials, 2005, 122(1/2): 119-128. |
| [11] |
Takahashi Y, Minamikawa R, Hattori K H, et al. Arsenic behavior in paddy fields during the cycle of flooded and non-flooded periods[J]. Environmental Science and Technology, 2004, 38(4): 1038-1044. DOI:10.1021/es034383n |
| [12] |
Arao T, Kawasaki A, Baba K, et al. Effects of water management on cadmium and arsenic accumulation and dimethylarsinic acid concentrations in Japanese rice[J]. Environmental Science and Technology, 2009, 43(24): 9361-9367. DOI:10.1021/es9022738 |
| [13] |
陈丽娜.不同水分管理模式下砷在土壤-水稻体系中的时空动态规律研究[D].保定: 河北农业大学, 2009. CHEN Li-na. Temporal and spatial dynamic of As in the soil-rice system under three water regimes[D]. Baoding: Hebei Agricultural University, 2009. |
| [14] |
Masscheleyn P H, Delaune R D, Patrick Jr W H. Effect of redox potential and pH on arsenic speciation and solubility in a contaminated soil[J]. Environmental Science and Technology, 1991, 25(8): 1414-1419. DOI:10.1021/es00020a008 |
| [15] |
Xu X Y, McGrath S P, Meharg A A, et al. Growing rice aerobically markedly decreases arsenic accumulation[J]. Environmental Science and Technology, 2008, 42(15): 5574-5579. DOI:10.1021/es800324u |
| [16] |
Duxbury J M, Panaullah G M, Zavala Y J, et al. Impact of use of Ascontaminated groundwater on soil As content and paddy rice production in Bangladesh[J]. Food and Fertilizer Technology Center. Tech Bull, 2009, 180: 1-11. |
| [17] |
Wenzel W W, Kirchbaumer N, Prohaska T, et al. Arsenic fractionation in soils using an improved sequential extraction procedure[J]. Analytica Chimica Acta, 2001, 436(2): 309-323. DOI:10.1016/S0003-2670(01)00924-2 |
| [18] |
Nielsen S S, Kjeldsen P, Hansen H-C-B, et al. Transformation of natural ferrihydrite aged in situ in As, Cr and Cu contaminated soil studied by reduction kinetics[J]. Applied Geochemistry, 2014, 51: 293-302. DOI:10.1016/j.apgeochem.2014.10.014 |
| [19] |
Cornell R M, Schwertmann U. The iron oxides:Structure, properties, reactions, occurrences and uses[M]. New Jersey: John Wiley & Sons, 2003.
|
| [20] |
Wang J, Zeng X, Zhang H, et al. Effect of exogenous phosphate on the lability and phytoavailability of arsenic in soils[J]. Chemosphere, 2018, 196: 540-547. DOI:10.1016/j.chemosphere.2017.12.191 |
| [21] |
Wang J J, Bai L Y, Zeng X B, et al. Assessment of arsenic availability in soils using the diffusive gradients in thin films(DGT)technique:A comparison study of DGT and classic extraction methods[J]. Environmental Science:Processes and Impacts, 2014, 16(10): 2355-2361. DOI:10.1039/C4EM00215F |
| [22] |
Zhang H, Davison W, Gadi R, et al. In situ measurement of dissolved phosphorus in natural waters using DGT[J]. Analytica Chimica Acta, 1998, 370(1): 29-38. DOI:10.1016/S0003-2670(98)00250-5 |
| [23] |
Luo J, Zhang H, Santner J, et al. Performance characteristics of diffusive gradients in thin films equipped with a binding gel layer containing precipitated ferrihydrite for measuring arsenic(Ⅴ), selenium (Ⅵ), vanadium(Ⅴ), and antimony(Ⅴ)[J]. Analytical Chemistry, 2010, 82(21): 8903-8909. DOI:10.1021/ac101676w |
| [24] |
Das S, Hendry M J, Essilfie-Dughan J. Transformation of two-line ferrihydrite to goethite and hematite as a function of pH and temperature[J]. Environmental Science and Technology, 2010, 45(1): 268-275. |
| [25] |
Schwertmann U, Murad E. Effect of pH on the formation of goethite and hematite from ferrihydrite[J]. Clays and Clay Minerals, 1983, 31(4): 277-284. DOI:10.1346/CCMN.1983.0310405 |
| [26] |
Andreeva D, Mitov I, Tabakova T, et al. Influence of iron(Ⅱ)on the transformation of ferrihydrite into goethite in acid medium[J]. Materials Chemistry and Physics, 1995, 41(2): 146-149. DOI:10.1016/0254-0584(95)01520-5 |
| [27] |
Hansel C M, Benner S G, Fendorf S. Competing Fe(Ⅱ)-induced mineralization pathways of ferrihydrite[J]. Environmental Science & Technology, 2005, 39(18): 7147-7153. DOI:10.1021/es050666z |
| [28] |
Yee N, Shaw S, Benning L G, et al. The rate of ferrihydrite transformation to goethite via the Fe(Ⅱ)pathway[J]. American Mineralogist, 2006, 91(1): 92-96. |
| [29] |
Boland D D, Collins R N, Glover C J, et al. An in situ quick-EXAFS and redox potential study of the Fe(Ⅱ)-catalysed transformation of ferrihydrite[J]. Colloids and Surfaces A:Physicochemical and Engineering Aspects, 2013, 435: 2-8. DOI:10.1016/j.colsurfa.2013.02.009 |
| [30] |
Straub K L, Benz M, Schink B. Iron metabolism in anoxic environments at near neutral pH[J]. FEMS Microbiology Ecology, 2001, 34(3): 181-186. DOI:10.1111/j.1574-6941.2001.tb00768.x |
| [31] |
Weber K A, Achenbach L A, Coates J D. Microorganisms pumping iron:Anaerobic microbial iron oxidation and reduction[J]. Nature Reviews Microbiology, 2006, 4(10): 752. DOI:10.1038/nrmicro1490 |
| [32] |
Lindsay W L, Schwab A P. The chemistry of iron in soils and its availability to plants[J]. Journal of Plant Nutrition, 1982, 5(4-7): 821-840. DOI:10.1080/01904168209363012 |
| [33] |
Wolf I, Russow R. Different pathways of formation of N2O, N2 and NO in black earth soil[J]. Soil Biology and Biochemistry, 2000, 32(2): 229-239. DOI:10.1016/S0038-0717(99)00151-0 |
| [34] |
Vodyanitskii Y N. Iron hydroxides in soils:A review of publications[J]. Eurasian Soil Science, 2010, 43(11): 1244-1254. DOI:10.1134/S1064229310110074 |
| [35] |
Jain A, Raven K P, Loeppert R H. Arsenite and arsenate adsorption on ferrihydrite:Surface charge reduction and net OH-release stoichiometry[J]. Environmental Science and Technology, 1999, 33(8): 1179-1184. DOI:10.1021/es980722e |
| [36] |
Jia Y, Demopoulos G P. Adsorption of arsenate onto ferrihydrite from aqueous solution:Influence of media(sulfate vs nitrate), added gypsum, and pH alteration[J]. Environmental Science and Technology, 2005, 39(24): 9523-9527. DOI:10.1021/es051432i |
| [37] |
吴萍萍.不同类型矿物和土壤对砷的吸附-解吸研究[D].北京: 中国农业科学院, 2011. WU Ping-ping. The study of different minerals and soils on the adsorption-desorption of arsenic[D]. Beijing: Chinese Academy of Agricultural Sciences, 2011. |
| [38] |
Nielsen S S, Petersen L R, Kjeldsen P, et al. Amendment of arsenic and chromium polluted soil from wood preservation by iron residues from water treatment[J]. Chemosphere, 2011, 84(4): 383-389. DOI:10.1016/j.chemosphere.2011.03.069 |
| [39] |
Sun X, Doner H E. Adsorption and oxidation of arsenite on goethite[J]. Soil Science, 1998, 163(4): 278-287. DOI:10.1097/00010694-199804000-00003 |
| [40] |
Ford R G. Rates of hydrous ferric oxide crystallization and the influence on coprecipitated arsenate[J]. Environmental Science and Technology, 2002, 36(11): 2459-2463. DOI:10.1021/es015768d |
| [41] |
Goldberg S, Johnston C T. Mechanisms of arsenic adsorption on amorphous oxicles evaluated using macroscopic measurements, ribrational spectroscopy, and surface complexation modeling[J]. Journal of colloid and Interface Science, 2001, 234(1): 204-216. DOI:10.1006/jcis.2000.7295 |
| [42] |
Al-Abed S R, Jegadeesan G, Purandare J, et al. Arsenic release from iron rich mineral processing waste:Influence of pH and redox potential[J]. Chemosphere, 2007, 66(4): 775-782. DOI:10.1016/j.chemosphere.2006.07.045 |