 2020, Vol. 39
2020, Vol. 39



2. 贵州大学农学院, 贵阳 550025
2. College of Agriculture, Guizhou University, Guiyang 550025, China
土壤微生物是陆地生态系统的重要组分,是土壤碳、氮、磷及其他元素生物地球化学循环的重要驱动力。以往许多研究已经证明,土壤微生物群落的数量、多样性和群落结构的变化能够显著影响生态系统过程的速率和功能的发挥[1-2],以及生态系统面对全球变化的稳定性[3]。因此,明确不同环境下土壤微生物群落的构建特征及其关键影响因子,是深入理解及预测生态系统物质、能量循环及生态系统功能响应全球变化速率及其机制的基础[4]。土地利用方式是影响土壤微生物群落特征的重要因素之一,不同土地利用方式,特别是自然植被和农业土地利用方式之间的差异对土壤微生物群落组成和活性的影响,在中国[5-7]及世界其他区域[8-11]均有报道。青藏高原是陆地生态系统的重要组成,其东南部林芝地区是雅鲁藏布江、怒江、澜沧江和金沙江等大江的上游水源涵养中心,是重要的森林资源区,也是生态脆弱环境区[12]。同时,林芝地区也是西藏粮食主要产区之一,该区域农业土地开发利用历史悠久且格局多样[13]。近年来由于该区域经济发展和人口增长压力的需求,农业用地面积逐年增加,这不仅给维持森林生态系统带来了极大挑战,而且可能对区域陆地生态系统物质循环及生态系统功能产生潜在影响。因此,研究藏东南林芝地区土壤微生物群落对农业土地利用方式的响应特征及其关键影响因素,有助于深入认识农业土地利用对该区域生态系统稳定性的影响,进而为该区域农、林业土地资源的合理利用和可持续管理提供科学依据。不同的土地利用方式能够通过影响输入到土壤中有机碳的数量和质量、土壤理化性质及土壤过程[14-16]等对土壤微生物群落产生显著的影响。许多研究表明,自然植被转变为农业土地利用方式(例如农田管理、放牧草地等)后,可能会带来地上初级生产力下降、凋落物及向地下分配有机碳数量降低以及土壤理化性质改变等一系列变化,从而导致微生物群落数量[9]、活性[17]以及多样性[7]的下降。但是,近年来对亚马孙的一系列研究却发现,农业土地利用方式(放牧草地)下土壤细菌的α-多样性显著高于自然植被(原始雨林)[10, 18]。这些迥异的结果表明农业土地利用方式对土壤微生物群落的影响并不唯一,可能取决于土地利用方式的类型、强度、持续时间等因素,并且具有区域特性。对于青藏高原的农业土地利用方式,目前研究多关注放牧等管理措施如何影响土壤微生物群落[19-20],但是对农田种植土壤中微生物群落的响应特征仍然认识不足。Xu等[21]利用高通量测序对藏东南地区不同土地利用方式下土壤中丛枝菌根真菌群落进行了分析,发现农田(小麦、青稞)土壤中丛枝菌根真菌的多样性显著低于自然草地,但和自然森林土壤无显著差别。而其他土壤微生物群落如何响应该区域农业土地利用方式,目前仍不清楚。此外,细菌和真菌在生长速率、碳源分解能力、能量周转等方面有明显区别[1],因此土壤细菌和真菌群落对农业土地利用方式的响应可能存在差异。Jiao等[7]在对黄土高原的研究发现,土壤细菌群落对土地利用方式变化(退耕还林)的响应比土壤真菌群落敏感。目前传统的微生物测定方法(例如微生物量碳氮、土壤酶活性等)无法将土壤微生物群落区分出不同种群,而磷脂脂肪酸(PLFA)谱图法和高通量测序方法可以根据生物标志物将土壤微生物划分为不同的种群,从而能够深入分析农业土地利用方式对土壤微生物群落数量、多样性及群落组成的影响。藏东南林芝地区自然植被(森林)转变为农业土地利用方式(例如农田种植、放牧草场)后,输入土壤的有机碳数量和质量、农田管理措施等因素以及土壤理化性质的改变可能对土壤微生物群落产生显著的影响。然而,不同土地利用方式下土壤微生物数量、多样性、群落组成和结构的异同,尤其是明确哪些土壤微生物类群对农业土地利用方式的响应最为敏感,以及导致土壤微生物群落发生变化的关键环境因素是什么,亟需进行深入分析。本研究采集藏东南林芝地区农田(小麦种植)及放牧草地这2种农业土地利用方式下的土壤样品,以自然森林土壤样品为对照,利用PLFA谱图法和高通量测序分析不同土地利用方式下土壤细菌和真菌数量、多样性和群落组成的差异性,并结合土壤理化指标揭示驱动土壤微生物群落差异的关键因素,以期阐明农业土地利用方式对藏东南地区土壤微生物的影响特征及其作用机制,为该区域农业土地资源合理利用提供科学依据。
1 材料与方法 1.1 采样区域概况本试验在青藏高原东南部林芝地区(26°52′~30° 40′ N,92°09′ ~98°47′ E)进行。该区平均海拔约为3000 m,年平均温度为7~16 ℃,大于10 ℃年积温为2272 ℃,年平均降水量为600~800 mm。林芝地区约有46%的面积为森林(2.64×106 hm2),占西藏全部森林面积的80%。草地面积为2.91×105 hm2,其中适合作放牧草场的面积约为5.25×104 hm2。林芝地区是西藏粮食重要产区之一,适宜农业耕作的土地面积为3×104 hm2。该区的传统农业耕作为休闲和单作,化肥投入少,机械化程度低,种植的粮食作物主要为小麦和青稞。
1.2 土壤样品采集土壤样品采集于2013年7月进行。采样点位于林芝地区布久乡卓木村,当地大部分农田耕作时间为50年以上,施用有机肥,不使用除虫剂。每年作物种植前进行一次翻耕处理,草地放牧,放牧强度为轻、中度。在典型农田(小麦种植)中随机选取5个样方(5 m×5 m),每个样方中随机采集3个表层土壤(0~20 cm)样品并混合为一个样品重复。同时,在采样区域内选取放牧草地和自然森林,以同样的采样方法进行土壤样品的采集,即3个土地利用方式处理(农田、放牧草地、自然森林),每个处理5次重复,一共采集15个土壤样品。所选取的森林是以高山松(Pinus densa- ta)、川滇高山栎(Quercus aquifolioides)和林芝云杉(Picea likiangensis var. linzhensis)为主要树种构成的温带针阔混交林次生林,林下小乔木和灌木包括花楸(Sorbus pohuashanensis)、小檗(Berbens thunbergii)、胡枝子(Lespedeza bicolor Turcz.)等。所选取放牧草地的主要物种包括蓼科(Polygonum)、紫菀(Aster)、苔草(Carex)、葛缕子(Carum carvi L.)、老鹳草(Geranium)、禾本科(Poaceae)、蛇莓(Duchesnea indica)、米口袋(Gueldenstaedtia verna)等。将采集的土壤样品挑出可见的石块、土壤动物和植物残体,过2 mm筛。用于DNA提取的土壤保存于-20 ℃,用于PLFA测定的土壤冻干后保存于-20 ℃,测定土壤矿质氮(NH4+ -N和NO3- -N)含量的土壤样品则保存于4 ℃冰箱。剩余测定土壤其他指标的样品风干并在室温下保存。
1.3 试验方法 1.3.1 土壤理化性质测定土壤pH用1 mol·L-1 KCl浸提(土水比为1:2.5)并用电位法测定。土壤有机碳(SOC)、总氮(TN)含量使用元素分析仪测定(EA1108,Carlo Erba,Turin,Ita- ly)。土壤有效磷(AP)使用0.5 mol·L-1 NaHCO3浸提-钼锑抗比色法测定。土壤NH4+-N和NO3--N含量用0.01 mol·L-1 CaCl2溶液浸提(土水比1:10),并用流动分析仪(TRAACS 2000,Bran and Luebbe,Norderstedt,Germany)测定,有效氮含量为两者之和。土壤团聚体分组使用湿筛法:首先将250、53、20 μm的筛子按次序放置于特制铁架上方铁桶内,并将100 g风干土壤样品(过2 mm筛)均匀放置于最上层的250 μm筛子上。向铁桶内加入2 L去离子水并浸泡5 min后,以振幅4 cm、频率35次·min-1持续振荡10 min。振荡结束后,将各级筛子上的土壤颗粒分别洗入铝盒中,105 ℃烘干,待铝盒内无明显积水后,再烘干24 h并称质量。粒径为250~2000 μm的土壤颗粒为大团聚体(Macroaggregate),粒径为20~53 μm和53~250 μm的土壤颗粒为微团聚体(Microaggregate)。
1.3.2 土壤微生物量测定(PLFA法)土壤中PLFAs的提取根据Frostegård等[22]的方法并稍作改进。首先将5 g冻干土壤样品用19 mL提取液(氯仿:甲醇:柠檬酸缓冲液=1:2:0.8,pH=4)提取其中的全部脂肪酸。将提取的脂肪酸样品加入硅胶固相萃取柱,分别用6 mL氯仿、6 mL丙酮和3 mL甲醇对硅胶萃取柱进行连续洗脱,PLFA组分被收集在甲醇相中。之后用1 mL 1:1的甲醇甲苯溶液和1 mL KOH甲醇溶液(0.2 mol·L-1)将PLFA组分进行甲基化,反应生成磷脂脂肪酸甲酯(FAME)。制备好的磷脂脂肪酸甲酯样品在上样前加入脂肪酸19:0作为内标,使用气相色谱仪测定脂肪酸含量,并用微生物鉴定系统MIDI Sherlock 6.1(MIDI Inc.,Newark,DE)标定每种脂肪酸的名称。
各种PLFA指示的微生物类群参考文献[22]。提取PLFAs的总量用来表征总的微生物量。革兰氏阳性细菌用同型(iso-)和异型(anteiso-)的支链饱和脂肪酸(i14:0、i15:0、a15:0、i16:0、i17:0、a17:0)表征;革兰氏阴性细菌用直链饱和脂肪酸(14:0、15:0、17:0)、单不饱和脂肪酸(16:1ω7c、18:1ω5c)以及环丙基脂肪酸(cy17:0、cy19:0)表征;放线菌用C10位置上有甲基支链的饱和脂肪酸(10Me16:0、10Me17:0、10Me18:0)表征;真菌用脂肪酸18:2ω6,9c和18:1ω 9c表征。
1.3.3 土壤微生物多样性及群落组成使用PowerSoil DNA Isolation Kit(MoBio Laborato- ries Inc.,Carlsbad,CA,USA)试剂盒提取土壤DNA,使用NanoDrop ND-2000分光光度计(Thermo Scientific,Wilmington,DE,USA)测定DNA的质量。使用引物515F(5′ -GTGCCAGCMGCCGCGG-3′)和907R(5′ - CCGTCAATTCMTTTRAGTTT-3′)对细菌16S rRNA基因进行扩增,使用引物1737F(5′ - GGAAGTA- AAAGTCGTAACAAGG-3′)和2043R(5′ - GCTGC- GTTCTTCATCGATGC-3′)对真菌ITS基因进行扩增,PCR产物纯化后送往上海美吉生物医药科技有限公司进行MiSeq高通量测序。测序获得的原始序列经过QIIME平台筛选和序列优化后,依据97%的相似度划分OTU(软件平台Usearch,version 7.0,http//drive.5.come/uparse/)。采用RDP classifier(verson 2.2,http://sourceforge.net/projects/rdp-classifier/)贝叶斯算法对OTU代表序列进行分类学分析,并在各个分类水平(门、纲、目、科、属、种)下统计分析各样本的群落组成,对比数据库为SILVA(Release 119,http://www.arb-silva.de)细菌和古菌16S rRNA基因数据库及UNITE(Release 6.0,http://unite.ut.ee/index.php)真菌ITS基因数据库。使用Mothur软件计算各个样本的α-多样性指数,其中丰富度用Chao1指数表征,多样性用香农多样性指数(Shannon-Weaver diversity)表征。
1.3.4 数据分析使用单因素方差分析(ANOVA)比较3种土地利用方式下土壤理化性质、土壤微生物量(PLFA含量)、土壤微生物α-多样性、细菌及真菌各种群相对丰度的差异,并用Duncan′s multiple range test检验各处理间差异的显著性。使用Pearson法分析土壤微生物量、多样性指数及各种群相对丰度与土壤理化指标的相关性。方差分析和相关性分析使用IBM SPSS Sta- tistics(version 22.0.0.0)进行。微生物群落结构及其影响因子使用基于Bray-Curtis距离矩阵的冗余分析(Distance-based redundancy analysis,db-RDA),3种土地利用方式下群落结构的差异用PERMANOVA进行检验,db-RDA分析使用R软件包vegan进行分析。利用随机森林模型(Random Forest)分析影响微生物群落在不同土地利用方式下构建差异的关键微生物种群及其贡献,每个物种的贡献用观测值和OOB(out-of-bag)预测值之间的均方误差增加值(Increase in MSE,%)表征。随机森林用R软件包randomForest进行运算分析,每个物种对预测模型贡献的显著性用软件包rfPermute计算。db-RDA分析及随机森林计算结果使用R软件包vegan及ggplot2进行绘制。
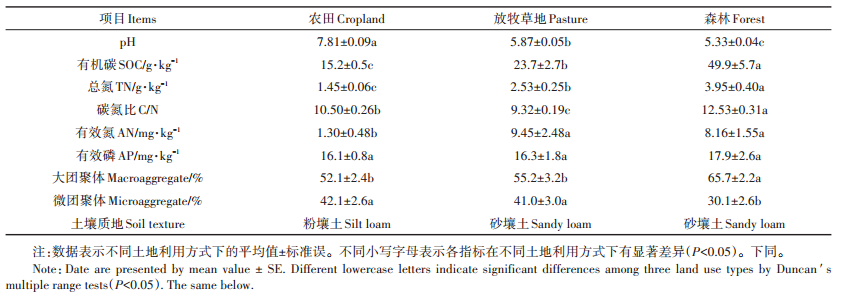
2 结果与分析 2.1 土地利用方式对土壤理化性质的影响农田耕作和放牧显著改变了土壤理化性质(表 1)。与自然森林相比,农田和放牧草地土壤的pH分别升高了2.48个和0.54个单位,SOC及TN含量分别降低了52.5%~69.5%和35.9%~63.3%。土壤AN含量,农田土壤(1.30±0.48 mg·kg-1)显著低于草地和森林土壤(8.16~9.45 mg·kg-1),而土地利用方式对土壤AP含量没有显著影响。此外,农田土地利用方式显著影响了土壤物理性质。与森林相比,农田和放牧草地土壤大团聚体数量降低了16.0%~20.7%,而微团聚体数量则增加了36.2%~39.9%。农田耕作增加了土壤黏粉粒含量,质地为粉壤土,而森林和放牧草地土壤为砂壤土。
2.2 土地利用方式对微生物群落生物量及α-多样性的影响表征土壤微生物群落生物量的PLFA含量在不同土地利用方式下有显著差异(表 2)。农田和放牧草地土壤中提取的PLFA总量相比于自然森林分别降低了51.8%和38.7%。对于各类微生物功能群,其在土壤中的微生物量也均表现为森林>农田、放牧草地。相比于自然森林,农田和放牧草地土壤中革兰氏阳性细菌、革兰氏阴性细菌、放线菌和真菌的生物量分别降低了26.1%~47.6%、40.0%~61.1%、44.1%~ 60.6%和5.2%~31.3%。
|
|
表 2 3种土地利用方式下土壤微生物群落PLFA含量及α-多样性指数 Table 2 PLFA concentrations and diversity indices of soil microbial community under different land use types |
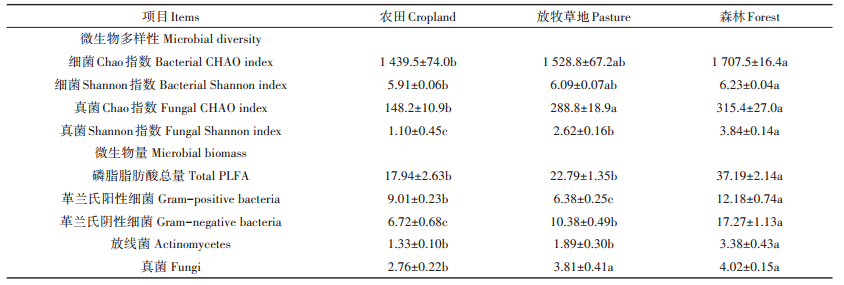
土壤细菌和真菌α-多样性指数(丰富度Chao、香农多样性)在不同土地利用方式下差异显著(P < 0.05),且均为森林>放牧草地>农田(表 2)。相比于自然森林,农田和放牧草地土壤中细菌群落的丰富度、香农多样性分别降低了10.5%~15.7%和2.2%~5.1%。土壤真菌群落多样性对农业土地利用的响应比细菌群落敏感,农田和放牧草地土壤中真菌丰富度和香农多样性比自然森林分别降低了8.4%~53.0%和31.8%~ 71.4%。
2.3 农业土地利用对微生物群落组成和结构的影响细菌群落中的主要种群在农田、放牧草地和自然森林土壤中的分布有明显差异(图 1)。农田土壤中放线菌门(Actinobacteria)的相对丰度(25.4%)显著高于放牧草地和自然森林土壤(17.6%~20.9%),然而浮霉菌门(Planctomycetes)、绿弯菌门(Chloroflexi)和厚壁菌门(Firmicutes)的相对丰度则相对于放牧草地和森林分别降低了33.1%~43.5%、32.9%~35.9%和43.3%~57.1%。拟杆菌门(Bacteroidetes)和蓝藻门(Cyanobacteria)在放牧草地土壤中的相对丰度(6.5%、6.1%)显著高于农田(3.1%、0.3%)和森林(3.8%、0.4%)土壤。其他优势细菌种群(在门水平上相对丰度大于1%),例如变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)对不同土地利用方式的响应不显著。

|
图 1 不同土地利用方式下土壤细菌和真菌群落组成 Figure 1 Composition of soil bacterial and fungal communities under different land use types |
与细菌群落相比,土壤真菌群落组成对土地利用方式的响应更加敏感(图 1)。子囊菌门(Ascomycota)是自然森林土壤真菌群落中的优势种群(相对丰度57.9%,主要的优势纲是粪壳菌纲Sordariomycetes),而农田耕作和放牧导致其在土壤中的相对丰度分别降低了89.6%和67.7%。农业土地利用显著提高土壤中担子菌门(Basidiomycota)(优势种群为伞菌纲Agaricomycetes)的相对丰度,其在农田(90.8%)和放牧草地土壤(75.9%)中的相对丰度分别是森林土壤(8.0%)的11.4倍和9.5倍。此外,农业土地利用导致接合菌门(Zygomycota)的相对丰度相较于自然森林(4.6%)显著降低了58.7%~67.4%(p < 0.05)。
基于距离矩阵的冗余分析(db-RDA)结果表明,不同土地利用方式下土壤细菌和真菌群落的结构有显著差异(P < 0.05)(图 2)。利用随机森林模型分析,进一步揭示了导致土壤细菌和真菌群落构建在3种土地利用方式下发生差异的关键微生物类群(细菌或真菌属)及其贡献。对于土壤细菌,显著影响(P < 0.05)3种土地利用方式下群落构建差异的细菌属主要为Actinoallomurus(Increase in MSE=2.20%)、Chtho- niobacter(1.99%)、Thermosporothrix(1.98%)、Mucilaginibacter(1.95%)等,并且这些种群基本都不属于群落中的优势种(在所有样品中的平均相对丰度都小于5%)(图 3)。而对于土壤真菌,显著影响(P < 0.05)3种土地利用方式下群落构建差异的真菌属主要为Inocybe(3.93%)、Emericella(2.93%)、Guehomyces(2.93%)、Wardomyces(2.52%)、Cylindrocarpon(2.52%)等,并且除了Wardomyces属在森林土壤中的相对丰度达到6%外,其他各属在土壤中的相对丰度均小于4%(图 4)。

|
图 2 不同土地利用方式下土壤细菌和真菌群落结构及其关键影响因子的冗余分析(基于距离矩阵) Figure 2 Differences in the structure of soil bacterial and fungal communities and the controlling factors among three land use types based on distance-based redundancy analyses |

|
左侧图表示各细菌属对不同土地利用方式下群落结构差异预测的贡献(Increase in MSE,%),右图表示对应的细菌属在各土地利用方式下的相对丰度 Figure in the left column present the predicted contribution(Increase in MSE, %)of indicator genus to the differences in bacterial community structure among three land use types. Figure in the right column present the relative abundance of each indicator genus under three land use types 图 3 不同土地利用方式下土壤细菌群落构建差异的指示种群(属水平)的随机森林预测分析 Figure 3 Results of random forest analyses that identifying the microbial indicators(at genus level)that can predict the differences in soil bacterial community among three land use types |

|
左侧图表示各真菌属对不同土地利用方式下群落结构差异预测的贡献(Increase in MSE,%),右图表示对应的真菌属在各土地利用方式下的相对丰度 Figure in the left column present the predicted contribution(Increase in MSE, %)of indicator genus to the differences in fungal community structure among three land use types. Figure in the right column present the relative abundance of each indicator genus under three land use types 图 4 不同土地利用方式下土壤真菌群落构建差异的指示种群(属水平)的随机森林预测分析 Figure 4 Results of random forest analyses that identifying the microbial indicators(at genus level)that can predict the differences in soil fungal community among three land use types |
相关性分析表明,土壤pH是影响土壤微生物量及多样性的关键环境因子(表 3)。土壤微生物生物量(革兰氏阴性细菌、放线菌、真菌PLFA含量及PLFA总量)、细菌和真菌群落的多样性指数(丰富度、香农多样性)均与土壤pH显著负相关(P < 0.05)。SOC和TN含量与微生物多样性指数、革兰氏阴性细菌PLFA含量有显著正相关关系,土壤有效氮含量与真菌PLFA含量有显著正相关关系(P < 0.05)。革兰氏阳性细菌PLFA含量显著受到土壤C/N的影响(P < 0.01)。
|
|
表 3 土壤微生物量、多样性指数及各种群相对丰度与土壤理化性质的相关性分析 Table 3 Pearson correlation coefficients(r)of soil microbial biomass, diversity index and relative abundance of various groups with soil general properties |
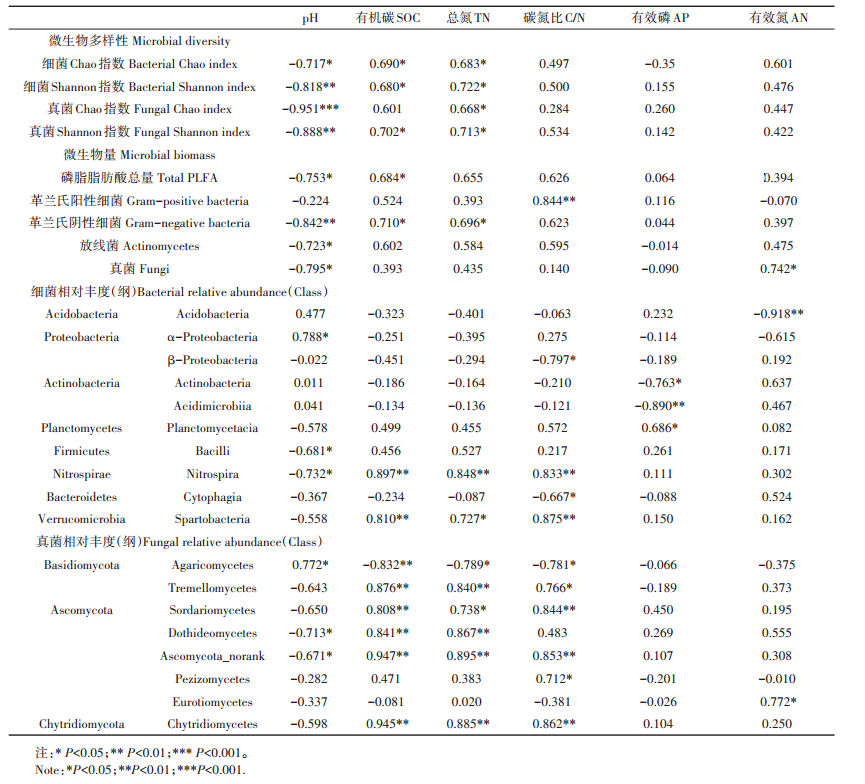
db-RDA分析表明,土壤pH、SOC、TN含量及土壤C/N是显著影响土壤细菌和真菌群落结构的关键环境因子(图 2)。在纲水平上,真菌群落中主要种群(平均相对丰度大于1%)的相对丰度在3种土地利用方式下的差异与4个关键环境因子显著相关(P < 0.05)(表 3)。Agaricomycetes的相对丰度与土壤pH正相关,与SOC、TN含量及C/N显著负相关,而其他纲(例如Sordariomycetes、Dothideomycetes、Tremello⁃ mycetes、Chytridiomycetes等)则与土壤pH负相关,与SOC、TN含量及C/N显著正相关。与真菌相比,细菌群落的主要纲与环境因子的相关性相对较弱。土壤pH显著影响α-Proteobacteria(正相关)、Bacilli和Ni- trospira(负相关)的相对丰度。Nitrospira和Spartobac⁃ teria的相对丰度与SOC、TN含量及土壤C/N显著正相关,而β-Proteobacteria和Cytophagia的相对丰度与土壤C/N显著负相关。此外,土壤AP含量显著影响Ac⁃ tinobacteria、Acidimicrobiia(负相关)和Planctomyceta⁃ cia(正相关)的相对丰度,而Acidobacteria的相对丰度则显著受到土壤AN的影响(P < 0.01)。
3 讨论自然植被转变为农业土地利用方式(农作物种植、放牧等),通常会导致一系列土壤环境因子,包括碳输入的数量和质量、土壤物理及化学性质等的剧烈变化[14-16],从而显著影响土壤微生物群落的数量、组成和活性[8-10]。本研究结果表明,在藏东南林芝地区,农业土地利用方式导致土壤微生物群落的总生物量较自然植被显著降低了38.7%~51.8%(表 2)。在其他区域或不同气候环境下,例如中国红壤地区[5]、加利福尼亚中部沿海地区[8]、肯尼亚西部[9]及热带雨林地区[11]等,也有研究报道了森林转变为农业利用后导致土壤微生物量下降的现象。本研究中,自然森林到农业土地利用方式的转变导致土壤pH增加了0.54~ 2.48个单位(表 1),而土壤pH的变化对土壤微生物量有关键的影响(表 3),尤其是对革兰氏阴性细菌(r= -0.842,P < 0.01)和真菌(r=-0.795,P < 0.05)。除了土壤pH,SOC和TN含量的增加显著提高革兰氏阴性细菌的生物量,这反映了革兰氏阴性细菌群落生存的“r-策略”:生长迅速,但碳源底物利用效率低(生长效率低),因而偏好碳源底物有效性高的环境[23]。与“r-策略”相反,“K-策略”的微生物,例如革兰氏阳性细菌,则能够将碳源储存在细胞内,消耗速率缓慢(碳源利用效率高),并且细胞壁富含肽聚糖且较厚,因此本研究结果发现革兰氏阳性细菌对土地利用方式转变导致的土壤pH及碳源有效性的响应并不显著,而只与土壤C/N显著正相关(r=0.844,P < 0.01)。真菌生物量和土壤AN含量有显著正相关关系(r=0.742,P < 0.05),可能是由于氮有效性的提高降低了其对真菌代谢的限制,真菌不需要分泌更多的胞外酶来分解有机质以获得氮源底物,因此更多的碳被分配到生物量构建而非胞外酶合成,从而导致真菌生物量增加。土壤微生物量是评价土壤肥力和质量的重要参数,因此农业土地利用情况下(农田和放牧草地)土壤微生物数量的剧烈下降说明在藏东南区域农业活动对土壤质量有负面影响。此外,土壤微生物量碳以及死亡后的残留成分(例如氨基糖)也是构成和维持土壤有机碳贮存的重要组分,农业土地利用方式下土壤微生物数量的下降从长期来看也不利于该区域土壤有机碳库的贮存和稳定。因此,如何在保障产量的前提下维持土壤微生物数量或者尽量减缓其降低,是该区域农业生产及土地利用需要进行探索的方向。
|
|
表 1 3种土地利用方式下土壤理化性质特征 Table 1 Soil general properties under three land use types |
以往对植物和动物群落的研究发现,人类活动造成的生态系统或环境的转变能够导致地上和地下生物群落多样性的降低[24]。本研究也得到类似的结果,即土壤细菌和真菌群落的α-多样性在自然森林下显著高于农业土地利用条件下(表 2)。Jiao等[7]在黄土高原退耕还林样地的研究也发现,还林20年以上的土壤细菌和真菌群落的α-多样性显著高于农田。然而在亚马孙雨林的一些研究却发现,自然森林转变为放牧草地后土壤细菌群落的α-多样性增加[10, 18]。土壤微生物群落多样性对森林-农业土地利用方式转变的不同响应很可能是由于不同区域地上、地下生产力差异而造成的。在上述提到的亚马孙地区的研究中,尽管热带雨林地上初级生产力高,但是地下初级生产力低,而转变为放牧草地极大提高了地下初级生产力[25],从而促进了土壤细菌群落α-多样性的增加。而在林芝地区,以高山松、林芝云杉和川滇高山栎为主要树种的针阔混交林下混有胡枝子、花楸等林下灌木,地上和地下初级生产力均高于农田和放牧草地,很可能是导致农业土地利用方式下土壤细菌和真菌群落的α-多样性显著降低的重要原因。此外,本研究发现,农业土地利用导致的土壤微生物多样性下降与土壤pH的升高有极显著的相关性(表 3)。最近对全球189个位点、7560余份土壤样品的分析表明,土壤pH是决定土壤细菌群落物种及功能多样性的关键环境因子,但是和土壤真菌多样性的相关性较弱[26]。然而在本研究中,真菌群落的丰富度对pH变化的响应最敏感(r=-0.951,P < 0.001)。这种结果差异,一方面说明了土壤微生物在局部区域的生物地理分布模式及其影响因子通常与全球模式无法完全吻合。另一方面,真菌的生物学特性可能限制其扩散条件,并且碳源底物类型也会限制某些真菌的生长,因而本研究观测到的真菌多样性和土壤pH的高相关性,很可能实质上是受到土地利用方式改变带来的植被变化、碳源输入质量和数量变化的影响,而并非土壤pH的直接调控作用。土壤微生物的生物多样性是其发挥生态系统功能的基础[4],对土壤碳、氮循环过程尤为重要。农业土地利用方式导致的土壤微生物多样性降低将如何影响藏东南区域土壤碳氮循环过程,是未来需要重点关注的科学问题。
除了微生物量和α-多样性,土壤微生物的群落组成和结构也受到土地利用方式的显著影响(图 1和图 2)。目前已有许多研究结果,例如在亚马孙地区的一系列研究发现,农田耕作、种植园开垦、放牧等农业土地利用是影响土壤微生物群落组成和结构的重要因素[10-11, 18, 27]。本研究中自然森林转变为农业利用后土壤微生物群落组成和结构的改变与土壤pH的增加、SOC和TN含量及土壤C/N的下降密切相关(图 2、表 3)。但不同土壤微生物种群对农业土地利用方式及其导致的环境因子变化的响应不一,这取决于不同微生物种群在生理及生态学特性上的差异。例如,变形菌门的许多细菌(包括α-Proteobacteria、β-Proteo- bacteria等)以及拟杆菌门的细菌多数是富营养型(Copiotrophic)[28]。我们发现农田土壤中α-Proteobac- teria的相对丰度(22.7%±1.7%)显著高于森林土壤(15.4%±0.2%,P < 0.001),很可能是由于有机肥的施用提高了农田土壤肥力。王慧颖等[29]在东北黑土区的研究也同样发现,长期施用有机肥和氮肥显著提高了α-Proteobacteria在土壤中的丰度。β-Proteobacte- ria和Cytophagia(属于拟杆菌门)在放牧草地土壤的相对丰度均显著(P < 0.01)高于森林土壤,并且与土壤C/N显著负相关,这与以往研究报道的这两类细菌偏好利用易分解碳源的结论一致[28]。相对于富营养菌,贫营养型(Oligotrophic)微生物,例如Acidobacteria通常在土壤养分贫瘠的环境中有相对高的丰度,这与我们观察到的Acidobacteria相对丰度随土壤AN含量升高而降低(r=-0.918,P < 0.01)相吻合。
与土壤细菌相比,不同土地利用方式下土壤真菌群落组成的变化更加剧烈(图 1),并且不同种群的相对丰度与土壤SOC、TN含量及C/N的相关性更高(表 3)。通常情况下细菌的生长和周转速率较高,对外界干扰的抗性和恢复性也可能较高[1],因而土壤细菌对农业土地利用方式的敏感性低于土壤真菌。此外,土壤细菌和真菌对碳源底物的分解能力和偏好不同,某些真菌对碳源底物类型有更严格的需求[1],这也能够解释土壤真菌群落对农业土地利用方式变化的响应更多地依赖于土壤碳源底物的有效性(数量及质量)。我们发现农业土地利用方式极大提高了担子菌门在土壤真菌群落中的优势度(图 1),尤其是Agaricomy- cetes,其在农田和放牧草地土壤中的相对丰度(90.4%,74.1%)分别是自然森林土壤(3.9%)的23.2倍和19倍。Agaricomycetes多为腐生真菌,在有机质丰富的环境中能够迅速增殖,因此有机肥及牲畜粪便能够为其提供生长代谢底物,从而促进其相对丰度的增加[29]。并且,Agaricomycetes对养分和能源的竞争优势可能强于其他菌群,因而随着长期的农田耕作及施肥,其他真菌种群逐渐消亡,从而导致真菌群落的物种多样性显著下降(表 1)。此外,本研究发现土壤AN含量显著影响子囊菌门散囊菌纲(Eurotiomycetes)真菌的相对丰度(r=0.772,P < 0.05),表明这类真菌对土壤无机氮的敏感性较高,但是从生理及生态学层面上如何解释这一结果目前并不十分清楚。尽管分子生物学技术(例如高通量测序)能够从基因层面对土壤微生物多样性、群落组成和构建提供更丰富的信息,但目前仍有相当一部分土壤微生物的生理及功能未能十分明确。在本研究中,利用随机森林模型分析,我们发现了多个在不同土地利用方式下土壤微生物群落构建差异显著的关键种群(图 3和图 4),而这些微生物区系的变化很可能带来生态系统碳氮循环过程的变化。例如,Bradyrhizobium(慢生根瘤菌属)是一类共生固氮细菌,同时也有研究发现它在森林土壤中是优势种群之一[30]。我们发现Bradyrhizobium在农田和森林土壤中的相对丰度显著高于草地,说明共生固氮过程很可能对农田和森林更为重要。此外,我们发现Flexibacter(屈挠杆菌属)在草地土壤中的相对丰度显著高于农田和森林,而该属中的某些种,例如Flexibacter canadensis能够分泌N2O还原酶从而进行反硝化作用[31],这反映出草地土壤反硝化过程速率或强度很可能与农田和森林土壤有所差异,需要深入研究。
作为西藏土地开发利用历史最为悠久的地区和重要粮食产区之一,林芝地区农业土地利用面积正在逐年增加[13]。本研究的结果表明,自然植被转变为农业土地利用(例如作物种植、放牧等)显著影响了土壤微生物的生物量、多样性、群落组成和结构,进而可能对该区域生物地球化学循环、生态系统过程和功能产生不可忽视的影响。并且,Marshall等[32]在研究草原恢复过程时发现,尽管草原恢复后其植被组成与原始草原相似,但土壤微生物群落却与原始土壤有显著差别。Jangid等[33]的研究则发现,土壤微生物群落结构对土地利用方式变化的响应主要是由土地利用历史所决定,地上部植被类型和土壤理化性质对其影响相对较小。这些结果说明土地利用方式,尤其是农业土地利用对土壤微生物群落的影响可能存在“滞后效应(Legacy effect)”,即农业土地利用方式对土壤微生物群落的影响很可能持续较长时间,即使植被已经恢复,土壤微生物群落的改变仍需要相当长一段时间才能恢复到原始状态[34]。相比于低海拔地区,西藏高海拔地区的生态系统通常对气候变化和人类活动的响应更加敏感。因此,藏东南地区农业土地利用方式造成的土壤微生物群落结构和功能的改变将如何影响该区域陆地生态系统碳氮循环,其需要多久才能恢复到原始状态,这是未来需要深入探讨的关键科学问题。
4 结论藏东南林芝地区农业土地利用方式(农田种植、草地放牧)通过影响土壤理化性质,进而显著影响了土壤微生物数量、多样性、群落组成及结构。相比于自然森林,农业土地利用方式显著降低了土壤中总PLFA含量及各微生物类群PLFA含量。农田和放牧草地土壤中的细菌群落多样性显著低于森林,主要是促进了放线菌门细菌的生长,而抑制了浮霉菌门、绿弯菌门及厚壁菌门细菌的生长。土壤真菌对土地利用方式变化的响应比细菌敏感,农业土地利用导致土壤中担子菌门伞菌纲Agaricomycetes的相对丰度增加了19~23倍,而其他真菌种群的生长受到抑制,进而导致真菌多样性的显著下降。农业土地利用方式显著影响土壤细菌和真菌群落的组成和结构,其变化主要受到土壤pH和碳源有效性驱动,并且各微生物种群响应农业土地利用方式的异同取决于其生理和生态学特性(例如富营养型和贫营养型)的差异。
| [1] |
Strickland M S, Rousk J. Considering fungal:Bacterial dominance in soils-Methods, controls, and ecosystem implications[J]. Soil Biology and Biochemistry, 2010, 42(9): 1385-1395. DOI:10.1016/j.soilbio.2010.05.007 |
| [2] |
Allison S D, Lu Y, Weihe C, et al. Microbial abundance and composition influence litter decomposition response to environmental change[J]. Ecology, 2013, 94(3): 714-725. DOI:10.1890/12-1243.1 |
| [3] |
Delgado-Baquerizo M, Eldridge D J, Ochoa V, et al. Soil microbial communities drive the resistance of ecosystem multifunctionality to global change in drylands across the globe[J]. Ecology Letters, 2017, 20(10): 1295-1305. DOI:10.1111/ele.12826 |
| [4] |
Bardgett R D, van der Putten W H. Belowground biodiversity and ecosystem functioning[J]. Nature, 2014, 515(7528): 505-511. DOI:10.1038/nature13855 |
| [5] |
Yao H, He Z, Wilson M J, et al. Microbial biomass and community structure in a sequence of soils with increasing fertility and changing land use[J]. Microbial Ecology, 2000, 40(3): 223-237. DOI:10.1007/s002480000053 |
| [6] |
毕明丽, 宇万太, 姜子绍, 等. 利用PLFA方法研究不同土地利用方式对潮棕壤微生物群落结构的影响[J]. 中国农业科学, 2010, 43(9): 1834-1842. BI Ming-li, YU Wan-tai, JIANG Zi-shao, et al. Study on the effects of different land use patterns on microbial community structure in aquic brown soil by utilizing PLFA method[J]. Scientia Agricultura Sinica, 2010, 43(9): 1834-1842. DOI:10.3864/j.issn.0578-1752.2010.09.009 |
| [7] |
Jiao S, Chen W M, Wang J L, et al. Soil microbiomes with distinct assemblies through vertical soil profiles drive the cycling of multiple nutrients in reforested ecosystems[J]. Microbiome, 2018, 6: 146. DOI:10.1186/s40168-018-0526-0 |
| [8] |
Steenwerth K L, Jackson L E, Calderon F J, et al. Soil microbial community composition and land use history in cultivated and grassland ecosystems of coastal California[J]. Soil Biology and Biochemistry, 2002, 34(11): 1599-1611. DOI:10.1016/S0038-0717(02)00144-X |
| [9] |
Bossio D A, Girvan M S, Verchot L, et al. Soil microbial community response to land use change in an agricultural landscape of Western Kenya[J]. Microbial Ecology, 2005, 49(1): 50-62. DOI:10.1007/s00248-003-0209-6 |
| [10] |
Rodrigues J L M, Pellizari V H, Mueller R, et al. Conversion of the Amazon rainforest to agriculture results in biotic homogenization of soil bacterial communities[J]. Proceedings of the National Academy of Sciences of the United States of America, 2013, 110(3): 988-993. DOI:10.1073/pnas.1220608110 |
| [11] |
Krashevska V, Klarner B, Widyastuti R, et al. Impact of tropical lowland rainforest conversion into rubber and oil palm plantations on soil microbial communities[J]. Biology and Fertility of Soils, 2015, 51(6): 697-705. DOI:10.1007/s00374-015-1021-4 |
| [12] |
廖晓勇, 陈治谏, 王海明, 等. 西藏土地利用综合分区[J]. 山地学报, 2009, 27(1): 96-101. LIAO Xiao-yong, CHEN Zhi-jian, WANG Hai-ming, et al. The research about comprehensive division of Tibet land utilization[J]. Journal of Mountain Science, 2009, 27(1): 96-101. DOI:10.3969/j.issn.1008-2786.2009.01.014 |
| [13] |
关卫星, 焦国成, 刘启勇, 等. 西藏一江两河地区农业耕作制度的现状与改革对策[J]. 西藏农业科技, 2012, 34(4): 44-48. GUAN Wei-xing, JIAO Guo-cheng, LIU Qi-yong, et al. Present situation and reform countermeasure on agricultural tillage system in the "YLN" region of Tibet[J]. Tibet Journal of Agricultural Sciences, 2012, 34(4): 44-48. DOI:10.3969/j.issn.1005-2925.2012.04.011 |
| [14] |
Celik I. Land-use effects on organic matter and physical properties of soil in a southern Mediterranean highland of Turkey[J]. Soil and Tillage Research, 2005, 83(2): 270-277. DOI:10.1016/j.still.2004.08.001 |
| [15] |
Wilson B R, Koen T B, Barnes P, et al. Soil carbon and related soil properties along a soil type and land-use intensity gradient, New South Wales, Australia[J]. Soil Use and Management, 2011, 27(4): 437-447. DOI:10.1111/j.1475-2743.2011.00357.x |
| [16] |
Beheshti A, Raiesi F, Golchin A. Soil properties, C fractions and their dynamics in land use conversion from native forests to croplands in northern Iran[J]. Agriculture Ecosystems and Environment, 2012, 148: 121-133. DOI:10.1016/j.agee.2011.12.001 |
| [17] |
朱珂, 王蕊, 李刚, 等. 土地利用方式变化对白浆土微生物生物量碳及代谢特征的影响[J]. 农业环境科学学报, 2018, 37(10): 2194-2201. ZHU Ke, WANG Rui, LI Gang, et al. The response of microbial biomass carbon and metabolic characteristics of albic soil to land use change[J]. Journal of Agro-Environment Science, 2018, 37(10): 2194-2201. DOI:10.11654/jaes.2017-1566 |
| [18] |
da C Jesus E, Marsh T L, Tiedje J M, et al. Changes in land use alter the structure of bacterial communities in Western Amazon soils[J]. The ISME Journal, 2009, 3(9): 1004-1011. DOI:10.1038/ismej.2009.47 |
| [19] |
孙欣, 汪诗平, 林巧燕, 等. 基于分子生态学网络探究西藏草地放牧对土壤微生物群落的影响[J]. 微生物学通报, 2015, 42(9): 1818-1831. SUN Xin, WANG Shi-ping, LIN Qiao-yan, et al. Molecular ecological network analyses revealing the effects of livestock grazing on soil microbial community in the Tibetan grassland[J]. Microbiology China, 2015, 42(9): 1818-1831. |
| [20] |
薛亚芳, 宗宁, 何念鹏, 等. 长期围封和自由放牧对高寒草甸土壤微生物群落结构及碳源代谢多样性的影响[J]. 应用生态学报, 2018, 29(8): 2705-2712. XUE Ya-fang, ZONG Ning, HE Nian-peng, et al. Influence of longterm enclosure and free grazing on soil microbial community structure and carbon metabolic diversity of alpine meadow[J]. Chinese Journal of Applied Ecology, 2018, 29(8): 2705-2712. |
| [21] |
Xu M, Li X L, Cai X B, et al. Land use alters arbuscular mycorrhizal fungal communities and their potential role in carbon sequestration on the Tibetan Plateau[J]. Scientific Reports, 2017, 7: 3067. DOI:10.1038/s41598-017-03248-0 |
| [22] |
Frostegård A, Bååth E. The use of phospholipid fatty acid analysis to estimate bacterial and fungal biomass in soil[J]. Biology and Fertility of Soils, 1996, 22(1/2): 59-65. |
| [23] |
Blagodatskaya E V, Blagodatsky S A, Anderson T H, et al. Priming effects in Chernozem induced by glucose and N in relation to microbial growth strategies[J]. Applied Soil Ecology, 2007, 37(1/2): 95-105. |
| [24] |
Soares B S, Nepstad D C, Curran L M, et al. Modelling conservation in the Amazon basin[J]. Nature, 2006, 440(7083): 520-523. DOI:10.1038/nature04389 |
| [25] |
Cerri C E P, Paustian K, Bernoux M, et al. Modeling changes in soil organic matter in Amazon forest to pasture conversion with the Century model[J]. Global Change Biology, 2004, 10(5): 815-832. DOI:10.1111/j.1365-2486.2004.00759.x |
| [26] |
Bahram M, Hildebrand F, Forslund S K, et al. Structure and function of the global topsoil microbiome[J]. Nature, 2018, 560(7717): 233-237. DOI:10.1038/s41586-018-0386-6 |
| [27] |
Mendes L W, Brossi M J D, Kuramae E E, et al. Land-use system shapes soil bacterial communities in Southeastern Amazon region[J]. Applied Soil Ecology, 2015, 95: 151-160. DOI:10.1016/j.apsoil.2015.06.005 |
| [28] |
Fierer N, Bradford M A, Jackson R B. Toward an ecological classification of soil bacteria[J]. Ecology, 2007, 88(6): 1354-1364. DOI:10.1890/05-1839 |
| [29] |
王慧颖, 徐明岗, 周宝库, 等. 黑土细菌及真菌群落对长期施肥响应的差异及其驱动因素[J]. 中国农业科学, 2018, 51(5): 914-925. WANG Hui-ying, XU Ming-gang, ZHOU Bao-ku, et al. Response and driving factors of bacterial and fungal community to long-term fertilization in black soil[J]. Scientia Agricultura Sinica, 2018, 51(5): 914-925. |
| [30] |
Vaninsberghe D, Maas K R, Cardenas E, et al. Non-symbiotic Bradyrhizobium ecotypes dominate North American forest soils[J]. The ISME Journal, 2015, 9(11): 2435-2441. DOI:10.1038/ismej.2015.54 |
| [31] |
Jones A M, Hollocher T C, Knowles R. Nitrous oxide reductase of Flexibacter canadensis:A unique membrane-bound enzyme[J]. FEMS Microbiology Letters, 1992, 92(2): 205-209. DOI:10.1111/j.1574-6968.1992.tb05260.x |
| [32] |
Marshall C B, Mclaren J R, Turkington R. Soil microbial communities resistant to changes in plant functional group composition[J]. Soil Biology and Biochemistry, 2011, 43(1): 78-85. DOI:10.1016/j.soilbio.2010.09.016 |
| [33] |
Jangid K, Williams M A, Franzluebbers A J, et al. Land-use history has a stronger impact on soil microbial community composition than aboveground vegetation and soil properties[J]. Soil Biology and Biochemistry, 2011, 43(10): 2184-2193. DOI:10.1016/j.soilbio.2011.06.022 |
| [34] |
Buckley D H, Schmidt T M. The structure of microbial communities in soil and the lasting impact of cultivation[J]. Microbial Ecology, 2001, 42(1): 11-21. DOI:10.1007/s002480000108 |