 2020, Vol. 39
2020, Vol. 39



2. 西华师范大学环境科学与工程学院, 四川 南充 637009;
3. 中国科学院生态环境研究中心, 北京 100085;
4. 西昌学院农业科学学院, 四川 西昌 615000
2. College of Environmental Science and Engineering, China West Normal University, Nanchong 637009, China;
3. Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China;
4. College of Agriculture Science, Xichang College, Xichang 615000, China
抗生素自20世纪被发现以来,为人类传染病的防治[1]、畜牧养殖中饲料转化率的提高[2]以及农业病虫害的防治[3]做出了重要贡献。农业领域抗生素的滥用问题已经引起了世界各国的关注,中国抗生素使用量约占世界一半,其中52%用于农业生产中[4]。抗生素的大量使用导致抗生素抗性细菌(Antibiotics resistance bacteria,ARB)和抗生素抗性基因(Antibiotic resistance genes,ARGs)的富集和传播,对生态环境和人类健康构成严重威胁[5]。同时,绝大多数ARGs位于微生物细胞中可移动遗传元件(Mobile genetic elements,MGES,如质粒和整合子等)上,使耐药性细菌能迅速蔓延,ARGs在不同细菌种群间的基因水平转移(Horizontal gene transfer,HGT),更是加剧了ARGs在不同环境介质中的传播,且存在ARGs通过食物链向人体迁移的潜在风险[6-7]。
农田土壤是ARGs的重要储存库,也是抗生素环境效应形成的主要场所[8]。农田土壤中的ARGs主要包括天然土壤环境中ARGs背景值,以及通过有机肥施用、污水灌溉等农业活动进入土壤环境的ARGs[9-10]。不同种类的ARGs在农田土壤中被检出,其中包括四环素类、多重耐药性类、β-内酰胺类、氨基糖苷类、磺胺类、氯霉素类、万古霉素类、大环内酯类-林可酰胺类-链阳性菌素B类(MLSB类)等[11-12]。不同类型的农田土壤由于其生物[11]和理化性质[13-15]、受人为干扰程度和类型[14]的差异,可能导致了农田土壤ARGs组成特征的差异。如前期研究表明农田土壤的ARGs分布特征与土壤pH、总有机质、总氮、总磷、重金属等理化性质都有显著相关性[13-15]。微生物群落多样性作为重要的生物学指标,也是ARGs分布差异的主要驱动因子[11]。在农田土壤中,很多研究表明施用有机肥显著提高了土壤中ARGs的丰度和多样性,同时也可能导致耐药性致病菌的传播[16-17]。因此,作为受人为干扰较为频繁的土壤环境,在大尺度上或区域水平探究农田土壤ARGs的分布规律及其驱动机制,既可以揭示不同区域的ARGs的污染特征,也为ARGs的产生途径研究提供数据支撑。
新的ARGs不断被发现[18],针对ARGs的引物越来越多,这也要求有更高通量、更快速和灵敏的检测技术来探究环境样品ARGs的多样性。近年来,美国Wafergen公司开发的高通量荧光定量PCR(Highthroughput qPCR,HT-qPCR)技术为快速定量检测大量样品提供了新途径,其可同时进行5 184个qPCR反应[19]。该技术已应用于环境中水体[20]、污泥[21]、土壤[17]、污水处理厂[22]和植物[23]等各类样品ARGs的检测,为本研究提供了技术支撑。
以往大多数的稻田土壤ARGs的污染现状研究主要集中在我国南方的水稻种植区,如江西和湖南等[15, 24],中国西部稻田种植区的土壤ARGs分布特征仍不清楚。四川盆地属于亚热带季风气候,降水丰富,为我国主要的水稻种植区之一[25]。因此,本文采用HT-qPCR技术,对四川省7个不同区域的稻田土壤ARGs的丰度和多样性进行研究。旨在揭示四川省稻田土壤的ARGs分布特征和规律;探究四川稻田土壤的潜在“指示ARGs”及影响ARGs分布格局的主导环境因子。为全面了解四川省稻田土壤的ARGs污染现状提供重要数据,并为农田土壤的潜在生物污染评估和制定ARGs防控措施提供理论支持。
1 材料与方法 1.1 采样点信息于2016年11月在四川省的7个水稻种植区域采集了土壤样品(图 1),分别是巴中市(BZ)、广安市(GA)、南充市(NC)、成都市(CD)、绵阳市(MY)、攀枝花市(PZH)、西昌市(XC)。7个采样区域均长期施用农家肥,其中包括发酵的农家粪肥、沼气废液或附近养殖场的发酵粪肥等。每个采样点采用网格布点法,在50 m×50 m范围内,分别采集3份表层土壤(0~10 cm),清除植物根系、石砾等,过2 mm孔径的不锈钢筛。将过筛后的土壤取部分自然风干后过0.90 mm孔径筛,用于土壤理化性质分析。另取一部分过2 mm孔径筛的土壤装入塑封袋中置于-20 ℃的冰箱以备后续分子生物学分析。

|
图 1 稻田土采样分布情况 Figure 1 Soil sampling site locations of the 7 paddy soils |
采用称重法测定稻田土含水量,采用pH计(PHS-3C,上海,中国)测量土壤pH值[m(土):V(水)= 1:2.5],采用全自动间断化学分析仪(CleverChem 200+,Hamburg,德国)测定土壤总氮、硝态氮和铵态氮。土壤总有机质由TOC分析仪(Elementar-TOC & Water Analysis,德国)测定。本试验采用悬浮液培养法,通过测定短期内土壤硝化速率来估算土壤的硝化潜势[26]。
1.3 土壤总DNA提取与荧光定量PCR分析总DNA提取使用FastDNA spin kit for soil(MP Biomedicals,美国)试剂盒,每个土壤3个重复。DNA浓度用NanoDrop分光光度计(NanoDrop 2000,德国)测定。提取的DNA置于-20 ℃的冰箱,待用后续分析。
实时荧光定量PCR扩增分析使用SYBR® Permix ExTaqTM试剂盒(Tli RNaseH Plus)在CFX96 Optical Real - time Detection PCR System荧光定量仪(BioRad Laboratories Inc.,美国)上进行。测定16S rRNA基因,氨氧化古菌(Ammonia-oxidizing archaea,AOA)和氨氧化细菌(Ammonia-oxidizing bacteria,AOB)的amoA基因的丰度[27]。实时荧光定量PCR标准曲线采用含有16S rRNA基因、AOA的amoA和AOB的amoA基因质粒,质粒标线按10倍梯度稀释,标准曲线质粒浓度的变化范围为103~109 copies·μL-1。定量PCR的反应体系为20 μL,包括1 μL DNA模板、10 μL SYBR© Premix EX TaqTM(Til RNaseH Plus)、正反向引物各0.1 μL和8.8 μL的灭菌蒸馏水。阴性对照用灭菌双蒸水代替样品。
1.4 高通量荧光定量PCR(HT-qPCR)使用Wafergen SmartChip Real-time PCR System荧光定量的反应平台(Warfergen Inc.,美国;Takara Bio,日本)对296个引物对进行高通量荧光定量PCR分析。其中,296个引物包括了283个ARGs(针对9类主要的抗生素:氨基糖苷类、β-内酰胺类、氯霉素类、MLSB类、磺胺类、四环素类、万古霉素类、多重耐药性类和其他类)、8个转座酶基因、4个整合酶-整合子基因和1个16S rRNA基因[21, 28]。为保证每个样品DNA的初始浓度一致,使用Qubit 3.0荧光计(ThermoFisher Scientific,USA)测定所有样品DNA的浓度,然后稀释至相同浓度。每个PCR体系的引物、样品DNA、酶等由SmartChip多样品纳米分配器(WaferGen Inc.,美国)分别加样到SmartChip芯片孔(Takara Bio,日本)中,SmartChip芯片共有5 184孔,可同时进行qPCR反应。随后将SmartChip芯片置于Smart Chip Cycler高通量qPCR仪(Takara Bio,日本)进行反应。HT-qPCR反应体系为100 nL,各试剂终浓度为:1 ×Light Cycler 480 SYBR Green Ⅰ Master Mix、1 μg· μL-1 BSA、5 ng·μL-1 DNA模板、1 μmol·L-1前置和后置引物,无核酸酶双蒸水。扩增反应条件如下:95 ℃预变性10 min;95 ℃变性30 s,60 ℃退火30 s,扩增40个循环[28]。所有qPCR均进行了两次以上的技术重复,以灭菌水代替样品作为阴性对照。根据Wafergen SmartChip Real-time PCR System的检测限和灵敏度,对数据的筛选标准为:(1)扩增效率在80%~ 120%;(2)循环临界值(Cycle Threshold,Ct)小于31;(3)3个技术重复至少2个得到扩增为有效扩增。
基因相对拷贝数和绝对丰度分别用公式(1)和公式(2)进行计算。根据Looft等[19]的方法,高通量荧光定量PCR基因的相对拷贝数计算公式为(1);根据Zhu等[28]的研究,绝对丰度的计算公式为公式(2)。

|
(1) |

|
(2) |
使用SPSS 20进行方差分析(ANOVA),P < 0.05被认为具有统计学意义。使用R软件和Gephi(0.9.2)软件进行ARGs网络分析[29-30]。ARGs和环境因子相关性冗余分析(Redundancy analysis,RDA)采用Canoco 4.5软件。用Origin 8.5制图。
2 结果与分析 2.1 不同稻田土壤中ARGs的类型多样性如图 2所示,7个稻田土中,一共检出166种不同的ARGs,6种转座子基因,3种整合子基因。平均每个土壤中检出71种ARGs,在CD土壤检出的ARGs个数最多,为84个,PZH土壤最少,为56个。7个稻田土中检出的MEGs的数量范围为4~7种,在MY土壤检出的MEGs个数最多。对7个稻田土ARGs的检出个数进行方差分析(ANOVA)检验发现,稻田土壤样品间有差异。MY、PZH和XC土壤的ARGs个数无显著差异(P > 0.05),但都显著高于其余土壤(P < 0.05)。7个稻田土中检出的166种ARGs亚型中,主要的ARGs种类为多重耐药性类(22.89%)、β -内酰胺类(16.87%)、氨基糖苷类(16.27%)、四环素类(13.86%)、MLSB类(13.85%)、万古霉素类(7.23%)、磺胺类(2.41%)、氯霉素类(1.20%)和其他类(5.42%)9类(图 2b)。根据不同引物所对应的耐药机制,7个稻田土中抗生素抗性机制主要为抗生素失活(42.17%)、抗生素外排泵(32.53%)和细胞核糖体保护(22.29%)(图 2c)。

|
不同小写字母代表样点间ARGs差异显著,不同大写字母代表样点间MGEs差异显著。下同 Different lowercase letters indicate significant differences among ARGs of samples, different uppercase letters indicate significant differences among MGEs of samples. The same below 图 2 7个稻田土中的ARGs个数、各类ARGs的检出个数和各类抗生素抗性机制比例 Figure 2 The detected numbers of ARGs in the 7 paddy soils, the resistance genes detected in all samples were classified based on the antibiotic to which they confer resistance, and the percentage of the resistance mechanism |
7个稻田土中ARGs的基因拷贝数,即绝对丰度,范围为9.55×108(GA)~2.83×1010(NC)copies·g-1(干质量)(图 3a)。NC土壤ARGs的绝对丰度极显著高于MY和NC土壤(P < 0.01)。CD和GA土壤ARGs的绝对丰度极显著低于MY、NC和XC土壤(P < 0.01)(图 3a)。7个稻田土中丰度最高的ARGs类型均为多重耐药类,占总丰度的26.26%~77.57%,约3.15× 108~ 7.49×109 copies·g-1(干质量),以NC土壤中丰度最高(图 3b);其次为MLSB类,占总丰度的1.74%~ 22.60%,约1.55×108~4.79×109 copies·g-1(干质量)(图 3b)。氨基糖苷类抗性基因在NC土壤中丰度最高,而且显著高于其他土壤(P < 0.05),是其他6个土壤的4.65~189.43倍。四环素类抗性基因的绝对丰度以NC土壤为最高,达2.46×109 copies·g-1(干质量)。万古霉素类抗性基因在7个土壤中都有检出,丰度约为8.86×107~4.32×109 copies g-1(干质量),以NC土壤中最高。7个稻田土中MGEs的总拷贝数检测到的范围为1.14×109~2.29×1010 copies·g-1(干质量),NC土壤丰度显著高于其他样品(P < 0.05)。MGEs和ARGs的总基因拷贝数呈极显著正相关(R=0.979,P < 0.001)(图 3a)。

|
图 3 7个稻田土中的各类ARGs绝对丰度与相对丰度及其平均绝对丰度与平均相对丰度比例 Figure 3 The absolute copy number and the normalized copy number of ARGs and the averaged absolute copy number proportion and the averaged normalized copy number proportion of different ARGs types in the 7 paddy soils |
为评估ARGs在整个细菌群落中的相对丰度及传播风险,将ARGs和MGEs丰度归一化到单个细菌水平,平均到每个细胞中的ARGs和MGEs的基因拷贝数(图 3c),即相对丰度。7个稻田土中ARGs的相对丰度范围为0.006~0.025 copies·cell-1,平均每个细胞有0.01个ARGs。NC土壤ARGs的相对丰度显著高于其他6个土壤(P < 0.01)。7个稻田土的主导ARGs的种类为多重耐药类(49.26%)、MLSB类(11.29%)和β-内酰胺类(10.87%),占总相对丰度的71.42%(图 3d)。不同种类ARGs相对丰度的差异性分析表明,XC土壤的β-内酰胺类和多重耐药性类抗性基因的相对丰度最高,都显著高于其他土壤(P < 0.05);GA和BZ土壤的四环素类抗性基因相对丰度显著高于其他土壤(P < 0.05);NC土壤的氯霉素类抗性基因相对丰度最高,而PZH土壤的磺胺类抗性基因丰度最高。7个土壤样品中,平均每个细胞中有0.008±0.004个MGEs。其中,XC土壤MGEs的相对丰度最高,为0.02 copies·cell-1,显著高于其他样品(P < 0.05)。ARGs和MGEs的总相对丰度呈显著正相关(R=0.882,P=0.009)(图 3c)。
2.3 不同土壤中ARGs和MEGs的组成比较分析基于稻田土ARGs的相对丰度的热图(图 4)表明,不同土壤样品中的ARGs组成有所差异,每种土壤都有其独有的ARGs类群(图 4中B~G分组模块)。7个土壤共有的主导ARGs类群分别为oprJ、oprD、acrA-04(多重耐药性类)、vanC-03(万古霉素类)、tetG-01(四环素类)、aacC(氨基糖苷类)、mphA-01(MLSB类)、blaCTX-M-04(β-内酰胺类)、pncA(其他类)。每个样品的主导ARGs均不同。如PZH土壤的主导ARGs为aacC、acrA - 04、acrA - 05、mphA-01和oprD,占其总相对丰度的34.99%,其中acrA-04基因丰度是其他土壤中该基因的1.48~19.45倍。CD土壤的主导ARGs是tet(37)、ampC_bla DHA、fabK和blaCTX-M-03,其中,tet(37)的相对丰度比XC土壤高出2 843.39倍。各样品中都检出其独有的ARGs,在XC土壤中检出16种,而且其中的mexF(多重耐药性类)和fox5(β-内酰胺类)为XC土壤的主导ARGs,丰度占其总相对丰度的82.21%。7个稻田土中均检出MGEs,如intI-1(clinic)、tnpA-04、intI-1LC、tnpA-02等。其中,intI-1(clinic)在7个样品中都被检出,相对丰度范围为0.003~0.011 copies·cell-1。

|
图 4 基于稻田土ARGs相对丰度的热图 Figure 4 Heatmap based on the normalized copy number of ARGs profiles of the 7 paddy soils |
基于ARGs和MEGs的相对丰度,分析不同ARGs和MEGs在7个稻田土中相对丰度变化的相关性(相关性系数r范围0.5~1),将显著相关(P < 0.05)的基因呈现于网络图中(图 5)。相连线的两个基因为显著正相关,线条越粗,表明相关性越强。图中的圆圈大小表示模块中所占比例大小,根据ARGs之间相关性强弱和相关的ARGs个数决定。使用Gephi-0.9.2软件计算与分析,该网络分析的模块系数为0.513,该相关性网络分析可分为4个模块。网络图共有64个节点和312条边(图 5)。模块Ⅰ、模块Ⅱ、模块Ⅲ、模块Ⅳ分别占28.12%、26.56%、26.56%、18.75%。blaCMY2-02(β-内酰胺类)、tetL -02(四环素类)、oleC(MLSB类)、aadA-01(氨基糖苷类)分别为模块Ⅰ、Ⅱ、Ⅲ、Ⅳ的枢纽中心。

|
图 5 稻田土ARGs和MGEs网络分析图 Figure 5 Network analysis depicting co-occurrence patterns among ARGs and MGEs of the 7 paddy soils |
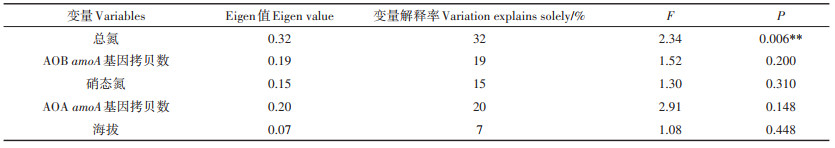
利用7个稻田土中相对丰度排在前50的ARGs和8种环境因子做冗余分析(图 6)。可见,7个样品分散于4个象限中,如CD和PZH土壤ARGs的多样性类似,均聚类在第2象限,而BZ和GA土壤的ARGs聚类在第3象限。影响土壤ARGs在不同地区分布格局的第1排序轴和第2排序轴特征值分别为42.6%、28.4%,可以解释ARGs差异总方差值的71.0%。冗余分析表明,总氮解释了32.0% ARGs多样性在不同土壤中的差异,对ARGs分布格局的影响达极显著水平(P=0.006)(表 1)。此外,土壤的硝态氮、pH、铵态氮、总有机质以及氨氧化微生物的丰度均不同程度地解释了ARGs多样性在不同土壤中的差异,但并无显著相关性(P > 0.05)。

|
图 6 基于相对丰度前50的ARGs和环境因子的冗余分析 Figure 6 Redundancy discrimination analysis(RDA)of paddy soil based on the normalized copy number of ARGs(top 50)and soil environmental factors |
|
|
表 1 基于相对丰度前50的ARGs和环境因子的冗余分析所得的Eigen、F和P值 Table 1 Eigen values, F values, P values obtained from normalized copy number of ARGs(top50)and redundancy analysis of soil environmental factors |
稻田作为一种典型的人为土壤,其生物理化性质受到自然与人为农业耕种因素的双重影响,在区域水平揭示稻田土壤ARGs多样性的分布格局,为理解农业活动对稻田生态系统中ARGs多样性的影响,以及为优化耕种和施肥方式提供依据。
四川省7个不同区域的稻田土ARGs的丰度和多样性有所差异。Tang等[15]在中国南方4个不同水稻试验田中也发现,不同样地的ARGs丰度和组成有显著差异,且ARGs丰度和组成受土壤理化性质、施肥频率的影响。土壤环境的差异会造成土壤微生物地理分布的不同[31],很多研究发现ARGs的多样性与微生物群落结构有显著相关性[21, 32],前期对这7个土壤的微生物指标研究发现其氨氧化微生物群落有显著差异[33],因此,推测微生物群落结构的差异可能导致这7个稻田土ARGs多样性地域分布差异。7个稻田土中ARGs的检出个数和丰度最高的均为多重耐药性类,与很多农田土壤中的研究结果类似[11, 34]。多重耐药性类抗性基因的传播对人类和动物的抗感染治疗是极大的威胁[35],一旦这些ARGs转移至致病菌中,则可能通过食物链或者直接感染进入人体,进而对当地农民的健康产生潜在威胁。如Chen等[36]研究表明施肥的土壤及其地上种植的植物叶际都检测到多种ARGs,且在植物叶际上大量检出在肥料中独有的ARGs。该研究表明,ARGs可能以土壤为载体迁移至植物或蔬菜叶际,进而进入食物链影响人类健康。因此,加强监管与教育,促进农用抗生素合理应用显得更为必要,尤其是对各类广谱抗菌药物的使用管制。万古霉素是人类抵抗细菌感染的“最后一道防线”之一[37],然而,随着在人类医疗中的广泛使用,万古霉素已在很多环境中富集,如河口沉积物[28]、土壤[34]、水体[21]。7个稻田土中万古霉素类抗性基因的检出表明该稻田生态系统可能受到人为污染源的影响。虽然这些ARGs可能仅存在于微生物中,实际不表现出抗性或在外界抗生素压力下才能表达[38],但对农田土壤中ARGs多样性的深入分析,能对ARGs潜在的农业环境风险做科学评估,同时为未来进一步检测农田土壤抗生素抗性指明了方向。此外,这些ARGs可能会随着土壤微生物通过孔隙水、雨水、灌溉水等载体,冲刷至周围的河流水体、池塘、井水或沉积物中[39],导致更大尺度的垂直或水平的ARGs基因转移和扩散,因此,农田土壤ARGs的富集所造成的潜在环境影响应高度重视。
7个稻田土中检出的ARGs基本涵盖了主要的已知抗生素类型,如氨基糖苷类(aac和aad等基因)、四环素类(tet基因)、β-内酰胺类(blaCTX-M和blaCMY等基因)等,这些ARGs类型在以往的土壤环境研究中也有检出[17, 40]。其中,7个稻田土中的主导ARGs之一aacC基因是在污水、废水和动物粪便中最频繁检出的一类ARGs[41],表明其可能是随施肥或灌溉等途径进入土壤环境。编码外排泵类四环素抗性基因tetG-01在7个土壤中有较高丰度,tetG基因在猪粪、土壤、沼气残渣、废水等样品中都被检出[13, 42],说明该四环素类抗性基因在环境中普遍存在,tetG基因随着粪肥施用和废水灌溉等农业活动进入农田土壤,并可能在农田土壤中富集。同时,抗生素的残留也是农田土壤ARGs富集的主要原因,如Zhu等[17]在我国多处大型养殖场的粪肥和周边土壤中发现,四环素的残留量可达15.2 mg·kg-1和0.78 mg·kg-1。且Gullberg等[43]发现即使是非常低浓度的抗生素浓度,也能对环境中的ARB和ARGs产生选择压力。通过施肥进入农田土壤环境的抗生素,与环境微生物产生互作效应,对环境中的土著微生物产生抗生素的选择压力和耐药基因的选择[44],本研究中的7个水稻种植点均施用有机肥,包括养殖场中发酵的粪肥,因此,不同土壤中的主导ARGs类型可能也受有机肥中残留抗生素的影响。如施用了当地养殖场粪肥的XC土壤中的mexF基因(多重耐药类)比在其他6个土壤中显著富集,这与黄福义等[45]的结果吻合,其发现施用了猪粪的水稻土壤中mexF基因的丰度相对于对照组土壤增加1 791倍。多重耐药类抗性基因acrA-04在PZH土壤中的显著富集(高达19.45倍),有研究表明acrA基因在养殖场的废水中[46]以及施用鸡粪的土壤中[11]也都有很高的检出率和丰度。因此,耕种、施肥、灌溉等农田管理措施,可能已造成了土壤中ARGs的富集,且四川省内不同地区的土壤具有明显的空间分布差异,未来进一步测定土壤抗生素残留情况及检测土壤抗性水平,对更深入地研究不同农业耕种区的ARGs污染状况有重要参考意义。
网络分析显示59种ARGs和5种MGEs的相对丰度之间存在关联(P < 0.05)。分析表明,每个模块的中心都可作为此模块中其他基因的“指示ARG”,模块中心的ARGs的微生物宿主可能与同一模块中的其他ARGs的宿主相同[47]。例如,blaCMY2-02是模块Ⅰ的中心,可作为在strB(氨基糖苷类)、blaOXY(β-内酰胺类)、aadA90-01(氨基糖苷类)等基因的“指示ARG”(图 5)。利用网络分析探索“指示ARGs”,可以快速估算环境样品中的ARGs污染状况。此外,许多研究表明ARGs在微生物间的传播和扩散与MEGs密切相关[17, 48],ARGs与转座子、整合子的显著正相关表明MEGs可能促进了ARGs在稻田土壤中的迁移和传播。本研究所有样品中均检测到整合子intI1(clinic),该基因被用作判定环境是否受人为源污染的标志基因[49]。研究发现医疗和环境菌株中的intI1与很多ARGs的基因盒相关,如氨基糖苷类抗性基因aadA、四环素类抗性基因tetG等都在intIl整合子基因盒中发现[50]。网络分析表明7个稻田土中的tetG、acrA-01、mexE等ARGs与intI1(clinic)、intI-1LC等整合子基因呈显著正相关(图 5),进一步表明稻田土壤中intI1整合子-基因盒系统对ARGs的传播起着重要作用,且基因盒可能含有多样性很高的ARGs。此外,基于ARGs和MEGs的相对丰度的网络分析可知,四环素类抗性基因tetL-2与转座子tnpA基因显著正相关(P < 0.05)(图 5),这些结果表明该基因可能存在于转座子上。如Chopra等[51]研究发现携带有tet基因的转座子将tet基因转座至不同的共轭质粒中。因此,ARGs的水平转移作为ARGs的重要传播方式应得到重视,并应加强农田土壤中ARGs的水平转移机制研究。
冗余分析表明,ARGs的组成和分布受多种理化性质的影响。总氮解释了32.0% ARGs组成在不同土壤中的差异,对ARGs分布格局的影响达极显著水平(P=0.006)(表 1)。这一结果与Cheng等[13]和成卫孝[52]在农田土壤中的研究结果一致。总氮是表征土壤肥力的重要指标之一,农田土壤中总氮的含量通常与施肥量呈正相关[53]。该结果说明施肥可能是这7个稻田土壤中ARGs的重要来源。因此,有必要针对性地制定科学合理的施肥制度,探究堆肥过程中耐药微生物和ARGs的产生与去除,以减少农田生态系统的生物污染。然而,土壤中ARGs的多样性受各环境因子的选择压力、基因水平转移、微生物增殖和死亡等多重因素的影响[17, 48, 54],仅依靠土壤理化性质可能难以精准预测或评估ARGs的来源和污染现状,因此,未来探究农田环境ARGs的污染机理和控制对策尤为重要。
4 结论(1)稻田土是ARGs的一个重要存储库,ARGs的水平转移可能促进抗性基因的迁移和富集,进而加剧农田土壤的ARGs污染。
(2)基于ARGs相关性的网络分析所识别的“指示ARGs”可作为评估四川稻田土ARGs污染水平的潜在方法。
(3)不同地区的稻田土壤ARGs组成各异,造成其差异的自然环境和人为因素,以及ARGs在农田环境中的传播机制仍需进一步探究。
(4)施肥可能是稻田土壤ARGs的重要来源,未来需加强对农田肥料,尤其是粪肥的ARGs污染风险评估。
| [1] |
Nigam A, Gupta D, Sharma A. Treatment of infectious disease:Beyond antibiotics[J]. Microbiological Research, 2014, 169(9): 643-651. |
| [2] |
PrzeniosÅ, osiwczynÂ′Ska M, Kwiatek K, et al. Use of antimicrobial agents in food production animals and the problem antimicrobial resistance[J]. Medycyna Weterynaryjna, 2015, 71(11): 663-669. |
| [3] |
朱昌雄, 宋渊. 我国农用抗生素的现状与发展趋势探讨[J]. 中国农业科技导报, 2006, 8(6): 17-19. ZHU Chang-xiong, SONG Yuan. Discussion on the situation and development of agro-antibiotic in China[J]. Review of China Agricultural Science and Technology, 2006, 8(6): 17-19. |
| [4] |
刘昌孝. 全球关注:重视抗生素发展与耐药风险的对策[J]. 中国抗生素杂志, 2019, 44(1): 1-8. LIU Chang-xiao. Global concern:Strategies for antibiotic development and risk of resistance[J]. Chinese Journal of Antibiotics, 2019, 44(1): 1-8. |
| [5] |
Knapp C W, Dolfing J, Ehlert P A, et al. Evidence of increasing antibiotic resistance gene abundances in archived soils since 1940[J]. Environmental Science & Technology, 2009, 44(2): 580-587. |
| [6] |
Hill E M. Decision making about antibiotic use:examining the role of antibiotic resistance knowledge, concern, and previous inappropriate antibiotic use[J]. Journal of Communication in Healthcare, 2017, 10(3): 226-233. |
| [7] |
Levy S B, Fitzgerald G B, Macone A B. Spread of antibiotic-resistant plasmids from chicken to chicken and from chicken to man[J]. Nature, 1976, 260(5546): 40. |
| [8] |
Hu H W, Wang J T, Li J, et al. Field-based evidence for copper contamination induced changes of antibiotic resistance in agricultural soils[J]. Environmental Microbiology, 2016, 18(11): 3896-3909. |
| [9] |
Tien Y C, Li B, Zhang T, et al. Impact of dairy manure pre-application treatment on manure composition, soil dynamics of antibiotic resistance genes, and abundance of antibiotic-resistance genes on vegetables at harvest[J]. Science of the Total Environment, 2017, 581-582: 32-39. |
| [10] |
Rizzo L, Manaia C, Merlin C, et al. Urban wastewater treatment plants as hotspots for antibiotic resistant bacteria and genes spread into the environment:A review[J]. Science of the Total Environment, 2013, 447(Complete): 345-360. |
| [11] |
Chen Q, An X, Li H, et al. Long-term field application of sewage sludge increases the abundance of antibiotic resistance genes in soil[J]. Environment International, 2016, 92-93: 1-10. |
| [12] |
Zhou X, Qiao M, Wang F H, et al. Use of commercial organic fertilizer increases the abundance of antibiotic resistance genes and antibiotics in soil[J]. Environmental Science and Pollution Research, 2017, 24(1): 701-710. |
| [13] |
Cheng W, Li J, Wu Y, et al. Behavior of antibiotics and antibiotic resistance genes in eco-agricultural system:A case study[J]. Journal of Hazardous Materials, 2015, 304: 18-25. |
| [14] |
Lin H, Sun W, Zhang Z, et al. Effects of manure and mineral fertilization strategies on soil antibiotic resistance gene levels and microbial community in a paddy-upland rotation system[J]. Environmental Pollution, 2016, 211: 332-337. |
| [15] |
Tang X, Lou C, Wang S, et al. Effects of long-term manure applications on the occurrence of antibiotics and antibiotic resistance genes (ARGs)in paddy soils:Evidence from four field experiments in south of China[J]. Soil Biology and Biochemistry, 2015, 90: 179-187. |
| [16] |
Jechalke S, Kopmann C, Rosendahl I, et al. Increased abundance and transferability of resistance genes after field application of manure from sulfadiazine-treated pigs[J]. Appl Environ Microbiol, 2013, 79(5): 1704-1711. |
| [17] |
Zhu Y G, Johnson T A, Su J Q, et al. Diverse and abundant antibiotic resistance genes in Chinese swine farms[J]. Proceedings of the National Academy of Sciences, 2013, 110(9): 3435-3440. |
| [18] |
Davis M A, Baker K N K, Orfe L H, et al. Discovery of a gene conferring multiple-aminoglycoside resistance in Escherichia coli[J]. Antimicrobial Agents and Chemotherapy, 2010, 54(6): 2666-2669. |
| [19] |
Looft T, Johnson T A, Allen H K, et al. In-feed antibiotic effects on the swine intestinal microbiome[J]. Proceedings of the National Academy of Sciences, 2012, 109(5): 1691-1696. |
| [20] |
Ouyang W Y, Huang F Y, Zhao Y, et al. Increased levels of antibiotic resistance in urban stream of Jiulongjiang River, China[J]. Applied Microbiology and Biotechnology, 2015, 99(13): 5697-5707. |
| [21] |
Su J Q, Wei B, Ou-Yang W Y, et al. Antibiotic resistome and its association with bacterial communities during sewage sludge composting[J]. Environmental Science & Technology, 2015, 49(12): 7356-7363. |
| [22] |
Pärnänen K M M, Narciso-da-Rocha C, Kneis D, et al. Antibiotic resistance in European wastewater treatment plants mirrors the pattern of clinical antibiotic resistance prevalence[J]. Science Advances, 2019, 5(3): eaau9124. |
| [23] |
Zhu B, Chen Q, Chen S, et al. Does organically produced lettuce harbor higher abundance of antibiotic resistance genes than conventionally produced?[J]. Environment International, 2016, 98: 152-159. |
| [24] |
Rahman M M, Shan J, Yang P, et al. Effects of long-term pig manure application on antibiotics, abundance of antibiotic resistance genes (ARGs), anammox and denitrification rates in paddy soils[J]. Environmental Pollution, 2018, 240: 368-377. |
| [25] |
梁涛, 陈轩敬, 赵亚南, 等. 四川盆地水稻产量对基础地力与施肥的响应[J]. 中国农业科学, 2015, 48(23): 4759-4768. LIANG Tao, CHEN Xuan-jing, ZHAO Ya-nan, et al. Response of rice yield to inherent soil productivity of paddies and fertilization in Sichuan basin[J]. Scientia Agricultura Sinica, 2015, 48(23): 4759-4768. |
| [26] |
路璐.亚热带酸性土壤中氨氧化微生物的生理生态过程研究[D].北京: 中国科学院大学, 2015: 32. LU Lu. The ecophysiological process of ammonia-oxidizing microorganism in subtropical acid soil[D]. Beijing: The University of Chinese Academy of Sciences, 2015: 32. |
| [27] |
Alam M S, Ren G D, Lu L, et al. Conversion of upland to paddy field specifically alters the community structure of archaeal ammonia oxidizers in an acid soil[J]. Biogeosciences, 2013, 10(8): 5739-5753. |
| [28] |
Zhu Y G, Zhao Y, Li B, et al. Continental-scale pollution of estuaries with antibiotic resistance genes[J]. Nature Microbiology, 2017, 2(4): 16270. |
| [29] |
Bastian M, Heymann S, Jacomy M. Gephi: An open source software for exploring and manipulating networks[C]//Third International AAAI Conference on Weblogs and Social Media, 2009: 341-364.
|
| [30] |
Fruchterman T M J, Reingold E M. Graph drawing by force-directed placement[J]. Software:Practice and Experience, 1991, 21(11): 1129-1164. |
| [31] |
Paul E A. Soil microbiology, ecology and biochemistry[M]. Academic Press, 2014.
|
| [32] |
Forsberg K J, Patel S, Gibson M K, et al. Bacterial phylogeny structures soil resistomes across habitats[J]. Nature, 2014, 509(7502): 612. |
| [33] |
Lu L, Li H L, He Y, et al. Diversification of ammonia-oxidizing microorganisms in seven geographically different paddy soils in Sichuan[J]. Journal of Chongqing University(English Edition), 2018, 17(4): 3-18. |
| [34] |
张毓森, 叶军, 苏建强. 粪肥与铜一次性施用对农田土壤抗生素抗性基因的长期影响[J]. 应用与环境生物学报, 2019, 25(2): 328-332. ZHANG Yu-sen, YE Jun, SU Jian-qiang. Long-term effect of onetime application of manure or copper antibiotic resistance genes in agricultural soil[J]. Chinese Journal of Applied and Environmental Bioligy, 2019, 25(2): 328-332. |
| [35] |
Oppegaard H, Steinum T M, Wasteson Y. Horizontal transfer of a multi-drug resistance plasmid between coliform bacteria of human and bovine origin in a farm environment[J]. Applied and Environmental Microbiology, 2001, 67(8): 3732-3734. |
| [36] |
Chen Q L, An X L, Zhu Y G, et al. Application of struvite alters the antibiotic resistome in soil, rhizosphere, and phyllosphere[J]. Environmental Science & Technology, 2017, 51(14): 8149-8157. |
| [37] |
Holmes A H, Moore L S P, Sundsfjord A, et al. Understanding the mechanisms and drivers of antimicrobial resistance[J]. The Lancet, 2016, 387(10014): 176-187. |
| [38] |
Arias C A, Murray B E. Antibiotic-resistant bugs in the 21st Century:A clinical super-challenge[J]. New England Journal of Medicine, 2009, 360(5): 439-443. |
| [39] |
Storteboom H, Arabi M, Davis J G, et al. Tracking antibiotic resistance genes in the South Platte River basin using molecular signatures of urban, agricultural, and pristine sources[J]. Environmental Science & Technology, 2010, 44(19): 7397-7404. |
| [40] |
Li J, Xin Z, Zhang Y, et al. Long-term manure application increased the levels of antibiotics and antibiotic resistance genes in a greenhouse soil[J]. Applied Soil Ecology, 2017, 121: 193-200. |
| [41] |
Mingeot-Leclercq M P, Glupczynski Y, Tulkens P M. Aminoglycosides:Activity and resistance[J]. Antimicrobial Agents and Chemotherapy, 1999, 43(4): 727-737. |
| [42] |
Liu M, Zhang Y, Yang M, et al. Abundance and distribution of tetracycline resistance genes and mobile elements in an oxytetracycline production wastewater treatment system[J]. Environmental Science & Technology, 2012, 46(14): 7551-7557. |
| [43] |
Gullberg E, Cao S, Berg O G, et al. Selection of resistant bacteria at very low antibiotic concentrations[J]. PLoS Pathogens, 2011, 7(7): e1002158. |
| [44] |
杨晓洪, 王娜, 叶波平. 畜禽养殖中的抗生素残留以及耐药菌和抗性基因研究进展[J]. 药物生物技术, 2014(6): 583-588. YANG Xiao-hong, WANG Na, YE Bo-ping. Review on antibiotic residues, antibiotic resistance bacteria and antibiotic resistant genes in livestock and poultry production[J]. Pharmaceutical Biotechnology, 2014(6): 583-588. |
| [45] |
黄福义, 李虎, 韦蓓, 等. 长期施用猪粪水稻土抗生素抗性基因污染研究[J]. 环境科学, 2014(10): 3869-3873. HUANG Fu-yi, LI Hu, WEI Bei, et al. Long-term manure application induced shift of diversity and abundance of antibiotic resistance genes in paddy soil[J]. Environmental Science, 2014(10): 3869-3873. |
| [46] |
Chen B, Hao L, Guo X, et al. Prevalence of antibiotic resistance genes of wastewater and surface water in livestock farms of Jiangsu Province, China[J]. Environmental Science and Pollution Research, 2015, 22(18): 13950-13959. |
| [47] |
Li B, Yang Y, Ma L, et al. Metagenomic and network analysis reveal wide distribution and co-occurrence of environmental antibiotic resistance genes[J]. The International Society for Microbial Ecology Journal, 2015, 9(11): 2490. |
| [48] |
Zhu Y G, Zhao Y, Zhu D, et al. Soil biota, antimicrobial resistance and planetary health[J]. Environment International, 2019, 131: 105059. |
| [49] |
Gillings M R, Gaze W H, Pruden A, et al. Using the class 1 integronintegrase gene as a proxy for anthropogenic pollution[J]. Isme Journal, 2014, 9(6): 1269-1279. |
| [50] |
Partridge S R, Tsafnat G, Coiera E, et al. Gene cassettes and cassette arrays in mobile resistance integrons[J]. FEMS Microbiology Reviews, 2009, 33(4): 757-784. |
| [51] |
Chopra I, Roberts M. Tetracycline antibiotics:Mode of action, applications, molecular biology, and epidemiology of bacterial resistance[J]. Microbiology and Molecular Biology Reviews, 2001, 65(2): 232-260. |
| [52] |
成卫孝.农业循环经济系统中抗生素和抗性基因的行为特征[D].杭州: 浙江大学, 2014: 45-48. CHENG Wei-xiao. The behavior of antibiotics and antibiotic resistance genes in circular agricultural system[D]. Hangzhou: Zhejiang University, 2014: 45-48. |
| [53] |
俞映倞, 薛利红, 杨林章. 不同氮肥管理模式对太湖流域稻田土壤氮素渗漏的影响[J]. 土壤学报, 2011(5): 988-995. YU Ying-liang, XUE Li-hong, YANG Lin-zhang. Effects of nitrogen management on nitrogen leaching of paddy soil in Taihu Lake region[J]. Acta Pedologica Sinica, 2011(5): 988-995. |
| [54] |
Vikesland P, Garner E, Gupta S, et al. Differential drivers of antimicrobial resistance across the world[J]. Accounts of Chemical Research, 2019, 52(4): 916-924. |